Market Access & Health Technology Assesment: Portugal




 Market Access & Health Technology Assesment: Portugal answers essential questions about this environment for pharmaceuticals in Portugal. It is a must-have asset for any company operating in Portugal or looking to enter the market.
Market Access & Health Technology Assesment: Portugal answers essential questions about this environment for pharmaceuticals in Portugal. It is a must-have asset for any company operating in Portugal or looking to enter the market.
Prepared in association with Cuatrecasas, one of the leading corporate law firms in Portugal.
October 2022
1. Expenditure Control and Cost-Contaiment Policies: Portugal
1. Please describe the main cost containment policies in place in your country and their fundamental principles
a. Pricing and impact of generic/biosimilar approval Currently there are no policies in place in this regard. b. Clawback/Payback/Discounts/Rebates In Portugal, the main mechanism for cost-containment is the prior evaluation, regulated by Decree-Law No. 97/2015 of June 1. The prior evaluation refers to prescription medicinal products that are intended to be purchased by hospitals and entities of the NHS. In practical terms, through the prior evaluation, Infarmed and pharmaceutical companies agree on a payback mechanism for each pharmaceutical product, i.e. on a maximum annual expenditure the Portuguese State is available to make for the use of a particular medicinal product; when such maximum annual expenditure is exceeded, pharmaceutical companies refund the Portuguese State, through the issue of credit notes in favour of the hospitals and entities of the National Health Service. c. Existence of Price/Volume agreements in the frame of public tendering Aside from the prior evaluation mechanism, within public tendering, and public procurement procedures in general, it is usual that pharmaceutical companies and public entities enter into commercial agreements, whereby the pharmaceutical company undertakes to grant to the public entity a discount/rebate when a certain volume of sales is reached. In Portugal, these agreements consist of commercial practices of market operators, but do not constitute governmental policies and they are not imposed to pharmaceutical companies. d. Existence of price freezes and cuts Not applicable. e. Post-launch monitoring of prescriptions/sales Not applicable. f. Existence of Generic Substitution Policies According to the dispensing rules in force in Portugal, pharmacists shall dispense generic medicinal products in substitution of original medicinal products, without prejudice to the customer's right to choose the original medicinal product. g. At prescriber level According to the prescription rules in force in Portugal, prescribers shall prescribe medicinal products based on the International Non-proprietary Name (INN) and they can only opt for prescription by the commercial name of the medicinal product or the marketing authorization holder in exceptional situations, namely when the original medicinal products does not have a reimbursed generic medicinal product. h. At retail level Not applicable.2. Are there any other policies in place aiming at cost control via incentive programs targeting the different actors (pharma companies, wholesalers, retailers, prescribers etc)?
There are no relevant policies to be highlighted.Also from this Market Access & Health Technology Assessment
2. Healthcare System and Funding: Portugal
1. Please make a general introduction to the public health sector in your country and its organization
The Public sector is composed of the National Health Service (Serviço Nacional de Saúde), also referred as the NHS, and the central and regional health services which integrate the NHS, amongst other relevant administrative companies and health services. The healthcare services provided by the healthcare establishments of the public sector tend to be free of charge to patients. These services are financed by government funds as defined in the national budget, compulsory tax contributions and user rates charged to patients - except for cases of waiver or exemption of payment of these rates.
Hospital medicine is free-of-charge for patients. Within the primary sector, patients have to pay part of the bill for prescription medicines at local pharmacies.
The companies placing the medicinal products on the Danish market are in principle free to set the prices for the medicinal products both within the primary sector and the secondary sector.
However, the unrestricted pricing is to a certain extent influenced by pricecap agreements negotiated every four years by the Ministry of Health, the Danish Regions, and the Danish Association of the Pharmaceutical Industry (Danish: Lægemiddelindustriforeningen or Lif). For more information on the price-cap agreements please see section III below.
2. Please provide any infographics including
a. The actors involved in the market access process (market authorization, pricing decisions, reimbursement decisions)
b. The information and data required

c. The process and flow
Submission > Pharmacotherapeutic Assessment > Economic Assessment > Negotiation > Recommendation > Decision
Also from this Market Access & Health Technology Assessment
3. Healthcare Actors and Payers: Portugal
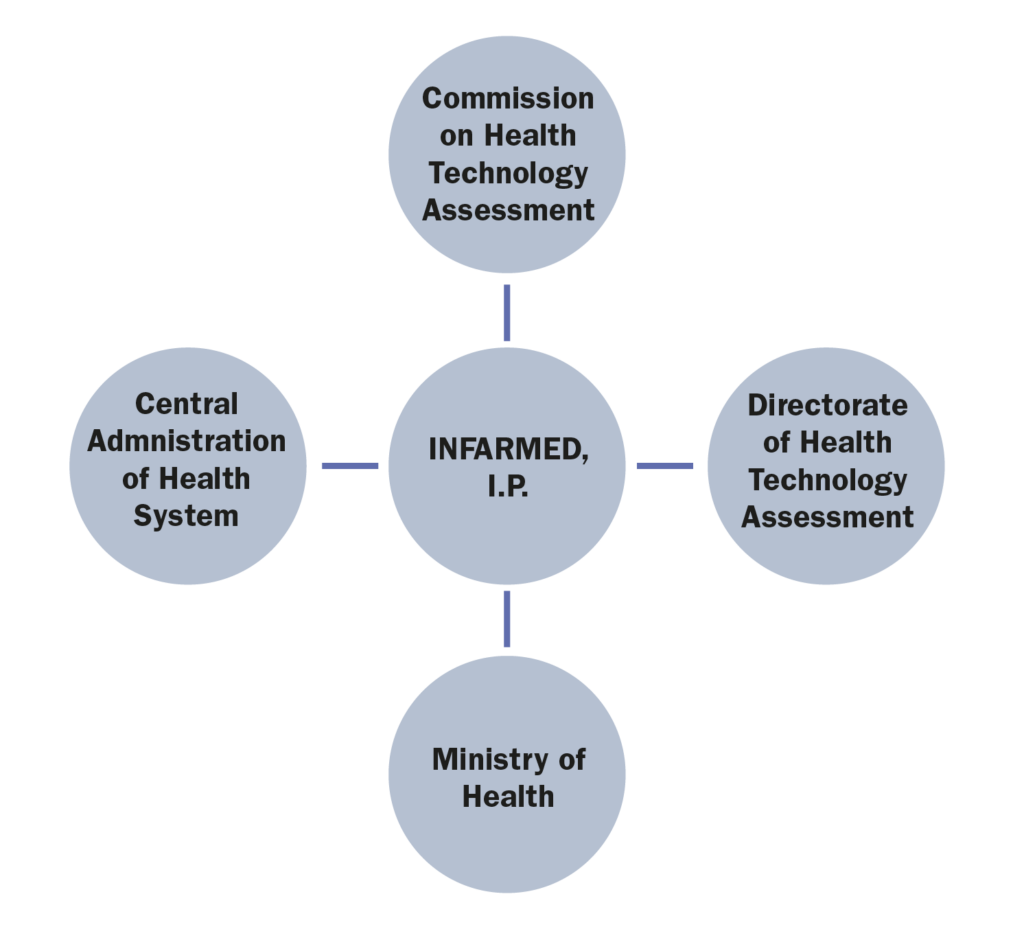
1.Which are the administrations, bodies and institutions in charge of public health in your country and what are their respective responsibilities?
The most relevant administrations, bodies and institutions in charge of public health in Portugal are:
• Ministry of Health (Ministério da Saúde), whose responsibility is, amongst others, to formulate, conduct, execute, and evaluate the national health policy;
• Directorate-General of Health (Direção Geral da Saúde), whose responsibilities are to plan, regulate, direct, coordinate and supervise all health promotion, disease prevention and healthcare activities, institutions and services, whether or not they are integrated into the NHS. It is also responsible for public health programs, quality and epidemiological surveillance, health statistics and studies;
• Central Administration of Health System (Administração Central do Sistema de Saúde), whose mission is to ensure the management of the financial and human resources of the Ministry of Health and the NHS, as well as the NHS facilities and equipment. In addition, it is responsible for defining and implementing policies, standards, regulations and integrated health planning, promoting innovation and efficiency in the NHS;
• Regional Health Administrations (Administrações Regionais de Saúde), which are responsible for implementing national health policy regionally, and coordinating all levels of healthcare.
• Executive Board of the National Health Service (Direção Executiva do Serviço Nacional de Saúde), entity recently incorporated that will coordinate and manage the assistance provided by the NHS, ensuring its functioning in a network, the continuous improvement of access to healthcare, the participation of users and the alignment of clinical and health governance.
2. Which are the administrations, bodies and institutions in charge of drug approvals in your country and what are their respective responsibilities?
Infarmed – National Authority of Medicines and Health Products, I.P (“Infarmed”), whose main goal is to ensure the quality, safety and efficacy of medicinal products and the quality, safety and performance of health products in order to avoid the risks of their use while ensuring adequate standards of public health and consumer’s protection.3. Which are the administrations, bodies and institutions that qualify as “payers” in your country and what are their respective responsibilities?
• The Government, whose mission is to ensure the management of human and financial resources of the Ministry of Health and the NHS, as well as its equipment and infrastructures;
• Insurance companies, whose responsibility is to provide access to medical care in the private sector;
• The public and private healthcare subsystems, such as ADSE, which guarantees the effective access to social protection regarding healthcare services to public administration workers and their families.
4. Which are the administrations, bodies and institutions in charge of pricing decisions in your country and what are their respective responsibilities?
Infarmed, whose mission is to regulate, evaluate, authorize, discipline, supervise, and ensure the surveillance and control of research, production, distribution, commercialization, and use of medicinal products for human use and health products.5. Which are the administrations, bodies and institutions in charge of reimbursement decisions in your countries and what are their respective responsibilities?
• Infarmed is also responsible for evaluating medicinal products for reimbursement purposes;
• The Ministry of Health is responsible for the reimbursement decision, although said power may be delegated to Infarmed.
6. Which are the administrations, bodies and institutions in charge of Health Technology Assessment in your countries and what are their respective responsibilities?
Health Technology Assessment is undertaken by two commissions of Infarmed, namely:
• The Commission on Health Technology Assessment (CATS) is an advisory body of Infarmed, composed of a pool of experts that are responsible for the assessment of the clinical and economic value of new health technologies, including medicinal products;
• The Directorate of Health Technology Assessment (DATS) is a department within Infarmed, responsible for managing the whole System of Assessment of Health Technologies (SiNATS), including the interaction between the marketing authorization holders, the CATS and the executive board of Infarmed.
7. Which are the administrations, bodies and institutions in charge of public procurement and tendering in your country and what are their respective responsibilities?
• Institute of Public Markets, Real Estate and Construction (IMPIC) is a public institute responsible for the supervision of public procurement, as well as public markets and construction and real estate sectors;
• The Court of Auditors (TC) is a supreme audit institution with several responsibilities. In regards public procurement matters, the Court of Auditors supervises public agreements and, in particular, is responsible for conducting a prior assessment and approval before some public agreements are executed, as these agreements represent a public expense;
• SPMS – Serviços Partilhados do Ministério da Saúde, which manages and centralizes public procurement procedures for some entities of the NHS.
8. What are the other actors of significance with regards to market access in your country and what are their respective responsibilities?
Infarmed and the Ministry of Health, as the entity responsible for the reimbursement decision.Also from this Market Access & Health Technology Assessment
4. Post Market-Approval Processes and Regulations: Portugal
1. What are the pricing models, processes and principles for originator drugs?
Prices of medicinal products are regulated by the System of Assessment of Health Technologies (SiNATS), approved by Decree-Law 97/2015, 1 June 2015, which established the provisions applicable to pricing, reimbursement, and prior evaluation procedures. The process for pricing decisions on originator drugs vary according to the type of medicinal product and intended market (outpatient vs. hospital and subject and not subject to medical prescription). For original medicinal products subject to a medical prescription provided in outpatient and in pharmacies, an international reference pricing system is applied to define the maximum market price. The international reference pricing system uses a basket of several EU countries, which are defined annually by the Ministry of Health. The reference countries selected for the year 2022 are Spain, France, Italy, and Slovenia. The maximum prices of original medicinal products subject to a medical prescription are subject to an annual revision, based on comparison with the prices approved in the reference countries. The prices of original medicinal products which require no prescription (OTCs) are free - unless they are reimbursed.2. What are the pricing models, processes and principles for generics and biosimilars drugs?
Generics subject to medical prescription are subject to specific pricing rules. The maximum sales price of generics is set by reference to the price of the reference medicinal product. The price of the generics cannot exceed 50% of the maximum sales price of the reference medicinal product or 25% of that price, should the reference product’s ex-factory price be lower than €10. Generics are also subject to an annual price revision. Generics which are not reimbursed and are sold to NHS Hospitals are also subject to a price approval and revision procedure. Under this regime, the price of the generic should be at least 30% lower than the price of the reference medicinal product. Biosimilar medicinal products are defined by exclusion, i.e. a biosimilar medicinal product is a biological medicinal product which does not fulfil the conditions of the definition of a generic (amongst other, because of differences related to the raw materials or to the manufacturing processes). Nevertheless, their regulation is similar to generic products in other regards. Biologic or biosimilar medicinal products do not follow a specific regime for the determination of their prices, therefore, if the biologic or biosimilar medicinal product is reimbursable under the applicable law, it may be subject to the regime of maximum prices as any other medicinal product.3. What are the reimbursement approval processes and principles for originator drugs?
There is a national reimbursement system of medicinal products that are provided outpatient and in pharmacies, in which the Government co-pays part of the price of the medicinal products prescribed to the users of the NHS. For a medicinal product to be reimbursed by the Government, the holder of the marketing authorization shall submit an authorization procedure for such purpose. There are four categories of reimbursement levels that range from a co-payment by the Portuguese State of 15% to 90%. In addition, there is also an exceptional reimbursement regime where some medicinal products duly specified may benefit from a 100% public financing.4. What are the reimbursement approval processes and principles for generics and biosimilar drugs?
Where a generic is placed in the market, a “homogenous group” (“HG”), composed of medicinal products with the same active substance and dosage, is created. Consequently, a reference price will be approved for the products which are part of said HG. The reference price will be based on the average of the retail sales price of the five lowest-priced medicinal products included in the HG. Following the approval of said reference price, the maximum amount of reimbursement for products included in the HG will be determined by applying the applicable reimbursement percentage to the reference price. The maximum sales price of generics placed in the market upon the HG creation must be at least 5% lower than the price of the cheapest generic already in the HG and has at least a 5% market share of generic medicinal products in the HG. Biosimilar medicinal products benefit from the reimbursement regime applicable to their reference biological medicinal product, without prejudice to any necessary adaptations. If the biosimilar medicinal product is part of a HG of biosimilar medicinal products, the maximum retail price of new biosimilar medicinal product to be reimbursed must be lower than the lowest priced biosimilar medicinal product, it being necessary that the lowest priced biosimilar medicinal product has a certain percentage of the market share of the medicinal products of the homogeneous group. Said percentage, as well as the margin for the maximum retail price, are defined by the government on a case-by-case basis. When the biosimilar medicinal product is not part of a HG, the maximum retail price of the biosimilar medicinal product may not be higher than 80% of the maximum retail price of the reference biologic medicinal product. Moreover, some biologic medicinal products are subject to exceptional regimes of reimbursements, where the reimbursement fee is of 100%. As for the prior evaluation agreements for biosimilar medicinal products, the economic advantage to be taken into account shall correspond to at least 20% of the direct price of the reference biological medicinal product. A biosimilar medicinal product shall benefit from the reimbursement system, either general or special, applicable to the reference biological medicinal product, by means of a reimbursement contract, with the adaptations resulting from the market share of both medicinal products and the prices of each one.Also from this Market Access & Health Technology Assessment
5. Price Control and Reference Pricing Systems: Portugal
1. Price
1.1. How does price control at ex-factory prices work in your country?
Ex-factory prices (PVA) are relevant to determine the retail price (PVP) of medicinal products and their maximum legal margins, as below.1.2. What are the pricing models, processes and principles for generics and biosimilars drugs?
The maximum price margin of medicinal products varies according to their ex-factory prices, as follows:- Ex-factory price equal to or less than €5:
- Wholesalers - 2.24 %, calculated over the ex-factory price, plus €0.25;
- Pharmacies – 5.58 %, calculated over the ex-factory price, plus €0.63;
- Ex-factory price equal to or less than €5:
- Wholesalers - 2.24 %, calculated over the ex-factory price, plus €0.25;
- Pharmacies – 5.58 %, calculated over the ex-factory price, plus €0.63;
- Ex-factory price from (euro) 5,01 to €7:
- Wholesalers - 2.17 %, calculated over the ex-factory price, plus €0.52;
- Pharmacies – 5.51 %, calculated over the ex-factory price, plus €1.31;
- Ex-factory price from €7,01 to €10:
- Wholesalers - 2.12 %, calculated over the ex-factory price, plus € 0.71;
- Pharmacies – 5.36 %, calculated over the ex-factory price, plus €1.79;
- Ex-factory price from (euro) 10,01 to €20:
- Wholesalers – 2.00 %, calculated over the ex-factory price, plus €1.12;
- Pharmacies – 5.05 %, calculated over the ex-factory price, plus €2.80;
- Ex-factory price from (euro) 20,01 to €50:
- Wholesalers – 1.84 %, calculated on the ex-factory price, plus €2.20;
- Pharmacies – 4.49 %, calculated on the ex-factory price, plus €5.32;
- Ex-factory price greater than €50:
- Wholesalers - 1.18 %, calculated on the ex-factory price, plus €3.68;
- Pharmacies – 2.66 %, calculated on the ex-factory price, plus €8.28.
1.3. How does price control at the retail pharmacy level work in your country?
Please refer to the question above.2. External Reference Pricing (ERP)
2.1. Is there a system of external reference pricing (ERP) in place in your country?
Yes.2.2. When and/or how often is ERP activated?
ERP is activated to define the maximum price of a prescription medicinal products, that was obtained a marketing authorisation, and that are provided outpatient and in pharmacies.2.3. What is the legal framework of the ERP in place in your country?
The ERP is governed by Decree Law n. º 97/2015 of 1 June 2015.2.4. What is the composition of the country basket?
The reference countries are defined annually by the Ministry of Health. The reference countries selected for the year 2022 are Spain, France, Italy, and Slovenia.2.5. Describe the price calculation and selection for reference products.
The retail prices of medicinal products to be placed for the first time in the Portuguese market shall not exceed the average resulting from the comparison with the prices in force in the reference countries for the same medicinal product. The retail price shall be calculated without taxes and duties applicable in the countries of reference, plus the commercialisation margins, taxes, and duties in force in Portugal. The retail price of generics corresponds to a percentage of the retail price authorized in Portugal for the reference medicinal product or, in the absence of such medicine, in the terms referred to original medicinal products.2.6. How often does the price need to be updated?
The prices of medicinal products are reviewed annually, based on a comparison with the prices charged in the reference countries. The medicinal products prices may also be exceptionally reviewed for reasons of public interest or at the initiative of the marketing authorization holder.2.7. How do the “price List”/catalogues from references countries work in your country?
Please refer to the previous questions.Also from this Market Access & Health Technology Assessment
6. HTA Decision Analysis Framework: Portugal
1. Which are the health technology assessment (HTA) evaluation bodies and their responsibilities in your country?
The CATS and the DATS are responsible for SiNATS.2. Do regulators require HTA studies in your country?
Yes, regulators may require HTA studies. Moreover, the information contained in SiNATS and the studies that support HTA decisions are published in accordance with the conditions to be defined by Infarmed.3. Do payers require HTA studies in your country?
Thought not mandatory, payers are allowed to require HTA studies in Portugal.4. How are HTA assessments translated into pricing conditions in your country?
HTA assessment is the basis for (i) deciding on the price, reimbursement, acquisition, or installation of health technologies and (ii) deciding on the maintenance of reimbursement or acquisition, through prior evaluation of health technologies.5. How are HTA assessments translated into reimbursement conditions in your country?
For the purposes of prior evaluation agreements and reimbursement matters, medicinal products must be subject to a pharmacoeconomic and pharmacotherapeutic evaluation.6. Which are the evaluation criteria, processes or models and analyses framework used for HTA in your country?
The assessment or re-evaluation is subject to a pharmacotherapeutic and/or economic evaluation. The technical-scientific criteria for assessing the different health technologies are established in a regulation approved by Infarmed.7. What is the methodology used in your country for HTA assessment?
HTA assessment aims to support the decision to use and finance health technologies in the NHS. This decision is based not only on the criteria of quality, safety, and efficacy but also on criteria of comparative economic effectiveness and efficiency, to optimize the use of available resources. Infarmed has published methodological guidelines for pharmacotherapeutic and economic assessment, as referred below.8. Which are the other decisions impacted by the assessed outcome in your country?
HTA assessment constitutes grounds to:- Decide on the price, reimbursement, acquisition or installation of health technologies;
- Issue recommendations or decisions on the use of any health technology;
- Decide on the maintenance of the reimbursement or the acquisition, after prior evaluation of health technologies.
9. Does your HTA review or inquire other international HTAs during the assessment process? If so, which ones are the usual partners?
Infarmed may enter into protocols or contract health technology assessment experts, whether legal or natural persons, national or foreign, to conduct health technology assessments.Also from this Market Access & Health Technology Assessment
7. HTA Dossiers: Portugal
1. Have local authorities published recommendations surrounding value assessment dossiers? (If yes please add link)
Yes. Infarmed has published methodological guidelines for pharmacotherapeutic and economic assessment. The recommendations can be consulted here.2. Have local Authorities published guidelines surrounding value assessment dossiers? (If yes please add link)
Yes. Please refer to Chapter VI, question 1.3. Have local authorities published official guidelines surrounding the submission of value assessment dossiers? (If yes please add a link)
Yes. Please refer to Chapter VI, question 1.4. Describe the overall process of preparing and submitting a HTA dossier in your country.
The process starts with the submission of the application, followed by the pharmacotherapeutic evaluation and the economic evaluation, where comparability is made through price-comparison, cost-minimization, and economic evaluation studies. Then, negotiation takes place, considering therapeutic alternatives, budgetary impact, and affordability. Finally, the recommendations and decisions are made.5. Describe the overall content of the HTA dossier in your country.
The HTA must contain technical information that demonstrates the medicinal product is safe, effective, and efficient for the claimed therapeutic indications.6. Which are the questions to focus on when preparing a HTA dossier in your country?
Please refer to the previous question.7. Which are the other strategic considerations to take into account when preparing a HTA dossier in your country?
There are several strategies that are worth following, namely:- The search strategy should be reproducible, established in line with the framework and the goal of the evaluation;
- Literature selection should be based on explicit inclusion and exclusion criteria, using standardized and recognized methodologies:
- Ineligible studies should be listed, along with the justification why they were excluded and a flow chart summarizing this information;
- Every effort should be made to include all relevant evidence, regardless of language.
Also from this Market Access & Health Technology Assessment
8. Data Requeriments: Portugal
1. In addition to the clinical data obtained through clinical studies please list the data required for:
a. Market approval The data that must be submitted varies according to the type of medicinal product and the applicable procedure. In any case, general requirements for obtainment of a marketing authorization apply, the applicant having to submit data such as:- General information of the medicinal product (proposed name, pharmaceutical form, quantitative and qualitative composition of all constituents, therapeutic indications, contraindications and adverse reactions, posology, method and route of administration, presentation and shelf life);
- Data concerning the manufacturing process and methods of the medicinal product;
- Description of the control methods employed by the manufacturer;
- Draft summary of product characteristics, product packaging mock-up, leaflet;
- Summary of the pharmacovigilance system,
- Risk management plan;
- Environmental risks report.