Market Access & Health Technology Assesment: Brazil





 Market Access & Health Technology Assesment: Brazil is a must-have asset for any company operating in Brazil or looking to enter the market.
Market Access & Health Technology Assesment: Brazil is a must-have asset for any company operating in Brazil or looking to enter the market.
Prepared in association with Trench Rossi e Watanabe, a leading Brazilian law firm.
Last Update April 2024
1. Healthcare System and Funding: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. Please make a general introduction to the public health sector in your country and its organization
As set out in the Brazilian Federal Constitution of 1988, health is a right of all and a duty of the state. It must be guaranteed by means of social and economic policies, with the purpose of reducing the risk of illness and other hazards. The Federal Constitution of 1988 considers the complementary existence of the private sector in health assistance. The public healthcare system was introduced by the Brazilian Federal Constitution of 1988 and is named the Unified Health System (SUS). SUS provides free and universal coverage and is funded through the social welfare budget of the Union, the states, the Federal District and the municipalities, as well as from other sources such as fines, fees and donations. The federal government, the states and the municipalities are responsible for the free distribution of medicines and medical devices. The Ministry of Health has a programme for the free distribution of essential and specialised medicines; the list of the medicines and health technologies covered is reviewed periodically. However, whenever the medicine is not available through the SUS system, individuals may file lawsuits against the government in order to force the government to purchase de product and deliver it to the specific individual (based on the argument that health is a right of all and a duty of the state). Such lawsuits are quite common and lead to discussions regarding their impact on government finances. The SUS also provides financial support to philanthropic and not-for-profit organisations, and to private health institutions by financial grants and reimbursement of medical procedures, devices and medicines upon the signature of an agreement between the private entity and the Ministry of Health. The reimbursement values are listed, along with the types of procedure and therapy that are covered. In the public sector, healthcare is delivered through programmes and plans that are implemented at federal, state and municipal levels. Actions and services must be organised and executed on a regional basis.2. Please provide any infographics including
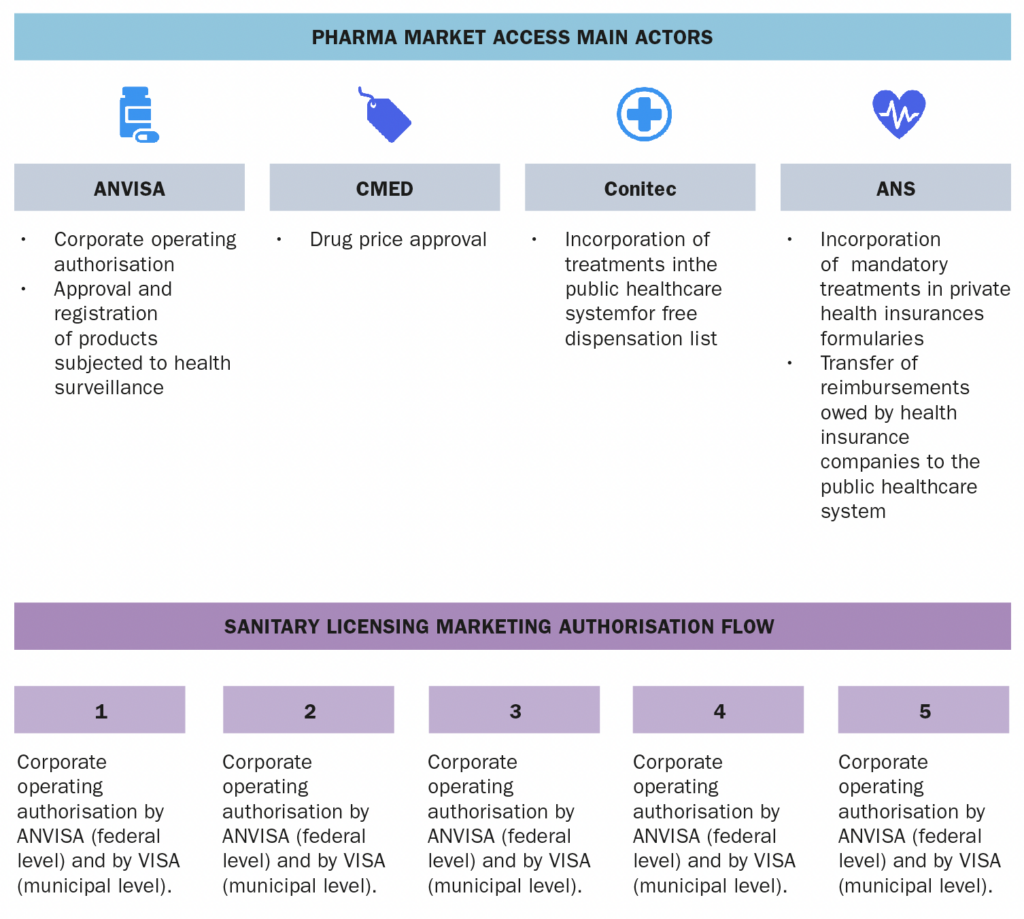
a. The actors involved in the market access process (market authorization, pricing decisions, reimbursement decisions) b. The information and data required c. The process and flow Also from this Market Access & Health Technology Assessment
2. Healthcare Actors and Payers: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. Which are the administrations, bodies and institutions in charge of public health in your country and what are their respective responsibilities?
SUS is managed collaboratively by the Federal Government, the States of the Federation, and the Municipalities. At the federal level, the SUS is managed by the Ministry of Health, which is responsible for formulating, regulating, inspecting, monitoring, and evaluating policies and actions, in coordination with the National Health Council. SUS also comprises, among others, the following public entities:- ANVISA – a regulatory agency responsible for the protection of public health by executing sanitary control over the production, importation, distribution, use and commercialization of a broad category of products such as drugs, medical devices, sanitizers, cosmetics, food, tobacco and services such as licensing conditions for health-related companies and healthcare institutions, among other attributions related to sanitary surveillance.
- Conitec – a multi-disciplinary collegiate formed by government representatives and civil society members, with advisory attributions related to the incorporation, exclusion or alteration of health technologies supplied by the SUS, as well as in the constitution of the protocols and guidelines for treatment in the public sector.
- CMED – entity responsible for regulating the pricing of drugs.
- ANS – a regulatory agency that regulates private health insurances.
2. Which are the administrations, bodies and institutions in charge of drug approvals in your country and what are their respective responsibilities?
The approval of drugs in Brazil is carried out by the National Health of Surveillance Agency (“ANVISA”), which is responsible for granting the product marketing authorisation after the evaluation of the manufacturing process, clinical trial results, stability, safety and efficacy data, as well as by Drug Market Regulation Chamber (“CMED”), which is the entity in charge of pharmaceutical market regulation and price approval of drugs.3. Which are the administrations, bodies and institutions that qualify as “payers” in your country and what are their respective responsibilities?
Organisms that qualify as payers are the federal, state and municipal entities that compose and manage the SUS, such as the Ministry of Health, health secretariats, public hospitals, public health foundations, etc. Health insurance companies, private hospitals, private health institutions, are also payers.4. Which are the administrations, bodies and institutions in charge of pricing decisions in your country and what are their respective responsibilities?
As mentioned in Question 2 above, CMED is the public entity responsible for pricing decisions. Among other attributions, CMED sets rules that stimulate competition in the sector, applies penalties when its rules are broken, as well as establishes and monitors the application of the mandatory minimum discount for public purchases.5. Which are the administrations, bodies and institutions in charge of reimbursement decisions in your countries and what are their respective responsibilities?
In the public sector, Conitec. In the private sector, ANS.6. Which are the administrations, bodies and institutions in charge of Health Technology Assessment in your countries and what are their respective responsibilities?
Health technology assessment is mainly conducted by Conitec, which is responsible for evaluating and recommending or not the incorporation of health technologies into SUS. Incorporation is implemented by SECTICS (the Secretariat of Science, Technology, Innovation and the Health Complex).7. Which are the administrations, bodies and institutions in charge of public procurement and tendering in your country and what are their respective responsibilities?
Each entity of public administration is able to carry out public procurement and tendering, in compliance with federal law, in order to purchase and hire. In the healthcare sector, procurement and tendering are usually conducted by the federal, state and municipal governments, as well as by public foundations, hospitals, etc. Note that, as a rule, all public purchases and hiring in Brazil must be preceded by a tendering procedure, except in cases of waiver and unfeasibility defined by law (for instance, sole source). Certain products are purchased directly by the Brazilian Ministry of Health, with the main purpose of having cost efficiency, economy and standardization.8. What are the other actors of significance with regards to market access in your country and what are their respective responsibilities?
The market access mainly involves the following public entities:- ANVISA, which is responsible for (at a federal level) authorising/licensing healthcare companies to operate in Brazil, and for granting product market authorisation.
- CMED, which is responsible for approving the drug prices.
- Conitec, which is responsible for recommending the incorporation of health technologies/drugs in the public health system, and
- ANS, which is responsible for regulating private health insurances and establishing the list of mandatory treatments that must be covered.
Also from this Market Access & Health Technology Assessment
3. Post Market-Approval Processes and Regulations: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. What are the pricing models, processes and principles for originator drugs?
To set the price of originator drugs, CMED uses Health Technology Assessment (“HTA”) to evaluate whether a drug provides an additional therapeutic benefit over comparators (which are drugs on the Brazilian market for the same indication). The assessment is based on the evidence submitted by the applicant, and a review of the technical literature carried out by the Executive Secretariat of the CMED (“SCMED”). In addition, to calculate the maximum price of originator drugs, CMED uses External Reference Pricing (“ERP”) and/or Internal Reference Pricing (“IRP”) criteria. The rules for price definition are currently provided for in CMED Resolution 02/2004.2. What are the pricing models, processes and principles for generics and biosimilars drugs?
To define the price of generic drugs, CMED uses the maximum ex-factory price (“PF”) of the reference drug. The PF of generic drugs must be 35% cheaper than the corresponding reference drug. As per CMED Notice 09/2016, regarding biosimilars drugs:- For those that prove therapeutic gain, the maximum PF cannot be higher than the lowest PF practiced in Australia, Canada, Spain, United States of America, France, Greece, Italy, New Zealand, Portugal, and in the country of origin of the product.
- For those that do not prove therapeutic gain and that are new to the list of drug products marketed by the company, the PF will be defined based on the average cost of treatment with drug products with a similar molecule, weighted by the turnover, and may in no case be higher than the lowest PF for the same product in the aforementioned countries.
- For those that do not prove therapeutic gain, but the company already has a product with a similar molecule on its list of marketed drug products, the PF will be defined based on the average cost of treatment of the drug products marketed by the company itself.
- For new presentations already marketed by the company itself with the same commercial brand, the PF will be defined based on the average cost of treatment with the same drug product.
3. What are the reimbursement approval processes and principles for originator drugs?
The reimbursement approval process for originator drugs (and for all other kinds of drugs) is conditioned to (i) their incorporation into SUS upon a favorable decision by Conitec, and (ii) their inclusion on the ANS list of treatments required for health insurance or their provision in the insurance contract itself. Nowadays, due to Law 14.307/2022, after being incorporated into SUS, the drug must also be included in the ANS list of mandatory treatments for private health insurances within 60 days.4. What are the reimbursement approval processes and principles for generics and biosimilar drugs?
The reimbursement approval process for generics and biosimilar drugs is the same as for the originator drugs, as mentioned in Question 3.Also from this Market Access & Health Technology Assessment
4. Price Control and Reference Pricing Systems: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. Price Control
1.1. How does price control at ex-factory prices work in your country? The Ex-Factory Price ("PF") is defined by CMED upon application by the company requesting drug marketing authorization, in accordance with the criteria established in CMED Resolution 02/2004. In Brazil, PF is the maximum price allowed for commercialisation of a drug to pharmacies, drugstores and public administration entities. CMED Resolution 02/2004 establishes 6 (six) different categories of drugs and specific rules for defining the PF of each of them. In brief, the drugs are classified into two clusters: categories I and II for new molecules, and categories III to VI for molecules already in the market. The CMED also defines the Maximum Price to the Government (PMVG), which is a mandatory discount over the PF, and applicable for sales to the Public Administration when the drug to be purchased is destined to comply with a court order or in cases where the drug is listed in the CMED Resolution 03/2011. In all other sales to the Government that do not fall into those situations, the company must observe at least the PF. The prices practiced in the market are monitored by CMED and the non-compliance to the PF constitutes an infraction subject to administrative sanctions. 1.2. How does price control at the wholesale level work in your country? At the wholesale level, PF also applies, as mentioned in Chapter 4, Question 1.1. 1.3. How does price control at the retail pharmacy level work in your country? At the retail pharmacy level, CMED regulates the Maximum Price for the Consumer (“PMC”), which is the maximum price allowed for commercialisation of a drug to the final consumer. PMC is defined in accordance with the rules set forth in CMED Resolution 02/2004. PMC is also monitored by CMED, and exceedance of the established limit leads to administrative sanctions.2. External Reference Pricing (ERP)
2.1. Is there a system of external reference pricing (ERP) in place in your country? Yes. CMED uses as reference for PF definition the ex-factory price practiced in Australia, Canada, Spain, United States of America, France, Greece, Italy, New Zealand, Portugal and in the country of origin of the product. 2.2. When and/or how often is ERP activated? ERP is applied in the analysis of the drug price application submitted to CMED, in case product is Category I, II or V, as per CMED Resolution No. 2/2004. 2.3. What is the legal framework of the ERP in place in your country? ERP is foreseen in CMED Resolution 02/2004, which provides on the criteria for the definition of drug prices. 2.4. What is the composition of the country basket? As mentioned in Chapter 4, Question 1.4., the country basket is composed by Australia, Canada, Spain, USA, France, Greece, Italy, New Zealand, Portugal and the ex-factory price practiced in the country of origin of the product. 2.5. Describe the price calculation and selection for reference products. Reference products are pricing in accordance with the rules foreseen in CMED Resolution 02/2004. Companies must submit to CMED the request for price approval, after receiving the marketing authorization from ANVISA. To calculate the maximum product price, companies must submit economic data on the product and propose a suggested price. Then CMED will define the maximum price of the drug, using ERP and/or IRP according to the category the product falls into. 2.6. How often does the price need to be updated? Prices are updated annually by CMED. The update is based on a price cap model calculated from an index, a productivity factor and an intra-sector and inter-sector relative price adjustment factor. 2.7. How do the “price List”/catalogues from references countries work in your country? As mentioned in Chapter 4, Question 2.2., the price catalogues from reference countries are used by CMED for the definition of the drug prices, in accordance with the rules provided in CMED Resolution 02/2004.3. Internal Reference Pricing (IRP)
3.1. Is there an internal reference pricing (IRP) system in your country? Yes. The CMED also uses the IRP as a parameter to define drug prices. 3.2. What is the legal framework of the IRP in place in your country? As well as ERP, IRP is foreseen in CMED Resolution 02/2004, which provides on the criteria for the definition of drug prices. 3.3. When and/or how often is IRP activated? IRP is applied for establishing the prices of generic drugs at 65% of the reference product.Also from this Market Access & Health Technology Assessment
5. HTA Dossiers: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. Have local authorities published recommendations surrounding value assessment dossiers? (If yes please add link)
Yes, but unfortunately only in Portuguese:2. Have local Authorities published guidelines surrounding value assessment dossiers? (If yes please add link)
Yes, but unfortunately only in Portuguese:- Proposal Application Guideline;
- Methodological Guidelines - Budgetary Impact Analysis - Manual for the Brazilian Health System;
- Methodological Guidelines - Systematic Review and Meta-Analysis of Diagnostic Accuracy Studies.
3. Have local authorities published official guidelines surrounding the submission of value assessment dossiers? (If yes please add a link)
Yes, but unfortunately only in Portuguese.4. Describe the overall process of preparing and submitting a HTA dossier in your country.
Regarding the preparation of the dossier, it is mandatory that the initial document be the letter properly addressed to DGITS/SECTICS/MS, containing identification of the applicant, the subject (name of the technology and indication requested), the place, date and signature. It is also required that the application form is completed accurately, printed, signed, and then included in the dossier. (Further details on the required documents are described in Question 5 below.) After gathering all the required documents, the dossier must be submitted to the Secretariat of Science, Technology, Innovation and Health Complex (“SECTICS”) through the Digital Register of the Ministry of Health, which can be accessed using this link. The system only allows files in pdf, html, xls and xlsx format, with up to 50MB.5. Describe the overall content of the HTA dossier in your country.
The dossier for requesting the incorporation of technology into SUS must contain: a. Letter: The letter must be on the first page of the process and must contain: identification of the applicant; identification of the recipient (DGITS/SECTICS/MS); subject (name of technology and indication requested); place, date and signature. b. Applicant documentation: For legal entities: articles of Incorporation of the company (notarized copy); and power of attorney of the applicant (in case the person responsible for signing the application is not included in the articles of incorporation). c. Application form: The application form must be filled out and submitted using this link. d. Main document: The document must contain: description of the disease/health condition related to the use of the technology; description of the technology; description of the scientific evidence of the technology compared to the one(s) already available at SUS (Systematic Review or Technical-Scientific Opinion); economic evaluation study from the perspective of SUS; budgetary impact analysis; bibliographic references; appendix (copy of package insert or instructions for use approved by ANVISA). e. Scientific studies d. Foreign language articles: Foreign articles not in English or Spanish must contain a sworn translation into Portuguese. e. Economic and budget impact studies: The studies must be available in .xlsm (Excel Macro Enabled Workbook File) or .trex (TreeAge) formats.6. Which are the questions to focus on when preparing a HTA dossier in your country?
The dossier should focus on demonstrating to Conitec the advantages of the HTA that justify its incorporation into the Brazilian public health system, such as its efficacy, safety and, especially, cost-benefit.7. Which are the other strategic considerations to take into account when preparing a HTA dossier in your country?
The same considerations mentioned in Question 6 above.Also from this Market Access & Health Technology Assessment
6. HTA Decision Analysis Framework: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. Which are the health technology assessment (HTA) evaluation bodies and their responsibilities in your country?
In Brazil, CONITEC is the main HTA evaluation body, which is responsible for recommending the incorporation of health technologies into the SUS, based on evidence of efficacy, safety and cost-benefits in relation to already existing technologies. However, ANVISA and CMED also act as HTA assessment bodies: the first evaluating the efficacy and safety during the process for granting the drug market authorization, and the second also evaluating evidence of efficacy and comparing treatment costs and international market to set the price of a drug. ANS is also an HTA assessment body, evaluating the mandatory coverage aspects.2. Do regulators require HTA studies in your country?
Yes. For incorporation purposes, as mentioned in item V.1 above.3. Do payers require HTA studies in your country?
As a rule, the entities that compose SUS only pay for technologies that have undergone HTA assessment by Conitec and that have been incorporated into the public health system. Private institutions and health insurances companies have the autonomy to purchase technologies not incorporated into SUS. However, the health technology must have been approved by ANVISA (since this is a legal requirement for marketing in Brazil).4. How are HTA assessments translated into pricing conditions in your country?
HTA is conducted after pricing is defined by CMED.5. How are HTA assessments translated into reimbursement conditions in your country?
Only health technologies that have undergone HTA evaluation by Conitec will be eligible for reimbursement since they must be incorporated into SUS — besides be covered by health insurances, which is an analysis and implementation conducted by ANS.6. Which are the evaluation criteria, processes or models and analyses framework used for HTA in your country?
The evaluation criteria applied in HTS are efficacy, effectiveness, safety, risks, costs, cost-effectiveness, cost-benefit and cost-utility, equity, ethics, economic and environmental implications.7. What is the methodology used in your country for HTA assessment?
There are four methodologies: Cost-Benefit, Cost-Minimization, Cost-Effectiveness and Cost-Utility. The most used approaches currently in the health sector are the last two. Cost-effectiveness analysis is a way of evaluating complete economic analysis in which both costs and consequences (outcomes) of health programs or treatments are examined. The result is expressed, for example, in cost per year of life gained. Cost-utility analysis is focused on the quality of the health outcome produced or avoided and introduces the QALY concept – quality-adjusted life years.8. Which are the other decisions impacted by the assessed outcome in your country?
Drugs included in the public health system after HTA assessment and recommendation by Conitec are compulsorily added to the ANS list, which establishes the mandatory treatments for private health insurances, due to Law 14.307/2022.9. Does your HTA review or inquire other international HTAs during the assessment process? If so, which ones are the usual partners?
Conitec requires that the assessment application be followed by international studies/articles on the technology to be evaluated.Also from this Market Access & Health Technology Assessment
7. Data Requirements: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. In addition to the clinical data obtained through clinical studies please list the data required for:
a. Market approval Each product category has its own data requirements, foreseen in specific regulations of ANVISA (which are constantly updated):- Resolution RDC 753/2022 provides for synthetic drugs, including generics and biosimilars.
- Resolution RDC 205/2017 – provides for orphan drugs.
- Resolution RDC 55/2010 – provides for biological drugs and biosimilars.
- Resolution RDC 721/2022 – provides for dynamized drugs.
- Resolution RDC 26/2014 – provides for phytotherapy drugs.
- Resolution RDC 73/2016 – provides for post-registration changes and cancellation of drug registration (this resolution is applicable for synthetic and semisynthetic drugs, including generics and biosimilars).
- Resolution RDC 505/2021 – defines the criteria to develop and to request the marketing authorisation of high technology products based on human cells and genes, called “advanced therapy medicinal products”.
Also from this Market Access & Health Technology Assessment
8. Managed Entry Agreements: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. Are there any Managed entry agreements in place in your country? (If so, please list them)
The first public and only managed access agreement reported in Brazil was signed between the Ministry of Health and Novartis in December 2022 for the incorporation of Zolgensma into SUS. The announcement of the Ministry of Health on the referred Agreement can be accessed in this link (unfortunately only in Portuguese). Such agreements have been used in the private sector.2. For each Individual Managed entry Agreement, describe the fundamentals of the Managed entry Agreement, its rationale and the process for implementing it.
There are no rules establishing the fundamentals for such contracts.3. When should this Managed Entry Agreement be considered?
We consider that in case of high cost products and when evidence with real life data is not well established yet.4. What is the potential impact on the product uptake?
N/A5. What are the potential challenges associated with this Managed Entry Agreement?
N/AAlso from this Market Access & Health Technology Assessment
9. Public Procurement and Tendering: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. Which are the main actors involved in public procurement and tendering?
As mentioned in Chapter 2, Question 7, each entity of public administration is able to carry out public procurement and tendering in order to purchase and hire. In the healthcare sector, procurement and tendering are usually conducted by the federal, state and municipal governments, as well as by public foundations, hospitals, etc.2. What are the main characteristics of the public procurement and tendering system?
The purpose of tendering is to achieve the proposal in the best interest of society and, at the same time, to ensure equality for all who wish to hire with the public administration. As a rule, all public purchases and hiring in Brazil must be preceded by a tendering procedure, except in cases of waiver and unfeasibility defined by law. Currently there are two public procurement and tendering laws in force: Law 14.133/2021 and Law 8.666/96 (which will be repealed at the end of 2023). Until the repeal of Law 8.666/96 occurs, the public administration entities may choose the law that will apply to their tendering procedures. The law chosen cannot be changed during the procedure/hiring and must be stated in the opening document. In contrast to other public administration entities, public enterprises and mixed economy companies follow the public procurement and tendering procedure established in Law 13.303/2016 ("State Enterprises Law").Also from this Market Access & Health Technology Assessment
10. Expenditure Control and Cost-Containment Policies: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. Please describe the main cost containment policies in place in your country and their fundamental principles
a. Pricing and impact of generic/biosimilar approval Generic drugs must have their PF 35% cheaper than the corresponding reference drug, as mentioned in Chapter 3, Question 2. b. Clawback/Payback/Discounts/Rebates As explained in Chapter 4, Question 1.1., PMVG is a mandatory discount over the PF, and applicable for sales to the public administration when the drug to be purchased is destined to comply with a court order or in cases where the drug is listed in the CMED's Resolution 03/2020. c. Existence of Price/Volume agreements in the frame of public tendering Although there is no legal obligation in this regard, public administration entities are allowed to enter into such agreements if relevant to pursue the best public interest. d. Existence of price freezes and cuts Due to the principle of legality foreseen in the Federal Constitution, price freezes and price reductions can only be determined by law and in justified circumstances. e. Post-launch monitoring of prescriptions/sales Whenever applicable, sales of drugs must respect the PF established by CMED, as well as the PMC at retail level, as explained in items Chapter 4, Question 1.1. and 1.3. f. Existence of Generic Substitution Policies SUS doctors must always prescribe the active ingredient of the drug instead of brand names, which promotes access to generics. g. At prescriber level The policy mentioned in Question 1 above. h. At retail level At the retail level, drugs must be sold respecting the PMC set by CMED, as detailed in Chapter 4, Question 1.3.2. Are there any other policies in place aiming at cost control via incentive programs targeting the different actors (pharma companies, wholesalers, retailers, prescribers etc)?
Yes. There are tax incentive programmes aimed at pharmaceutical companies and certain drugs receive tax incentives. There is also the Popular Pharmacy Programme, which offers free medicines or at significant discounts through partnerships with private pharmacies and drugstores. In addition, the SUS provides financial incentive programmes for private health institutions that work in partnership with the public health system, as well as for other health actions, depending on the relevance/necessity.Also from this Market Access & Health Technology Assessment
11. Essential Drug List: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. Has an essential or priority drug list been published in your country? (If so provide the link)
Yes. In Brazil, the National List of Essential Drugs (“RENAME”) provides the selection and standardisation of drugs indicated for treatment in the SUS. RENAME is updated every 2 years. The latest one published (in 2022) can be accessed using this link.2. If so what is the impact/consequences of a drug being published on the EDL/PDL:
a. In terms of market approval There is no impact on market approval. Note, however, that only drugs approved by ANVISA could be included in RENAME. b. In terms of reimbursement When included in RENEME, it means that drug has been incorporated into SUS. Therefore, if that drug is also included in the ANS List (which establishes the mandatory treatments for private health insurances) or provided for in the health insurance contract, whenever an insured receives such drug at SUS, the health insurance company will be obligated to reimburse the ANS, as detailed in Chapter 2, Question 5. As explained in Chapter 3, Question 3, nowadays, due to Law 14.307/2022, all drugs incorporated into the SUS must also be included in the ANS List within 60 days. c. In terms of pricing There is no direct impact on price. Drugs must be sold to SUS always respecting the PF defined by CMED. In case the drug purchase is destined to comply with a court order or is listed in CMED Resolution 03/2020, the drug should be sold respecting the PMVG, as explained in Chapter 4, Question 1.1. d. In terms of value assessment and pharmacoeconomic requirements There is no direct impact on value assessment and pharmacoeconomic requirements.3. Do value assessment and pharmacoeconomic data play a role in terms of access to the list?
Yes. The incorporation of the drug in SUS and inclusion in RENAME depends on a favourable decision by Conitec, which assesses pharmacoeconomic aspects of the ATH to verify whether its insertion in the public health system is pertinent.Also from this Market Access & Health Technology Assessment
12. Orphan Drugs: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. What is the definition of an orphan drug in your country?
There is no specific definition for orphan drug in Brazilian current rules. However, we understand that it corresponds with the definition of rare diseases. As per ANVISA Resolution RDC 205/2017, rare diseases are defined as diseases with incidence of up to 65 cases for every 100,000 inhabitants.2. Describe the pricing process of orphan drugs in your country.
The pricing of orphan drugs follows the rules foreseen in CMED Resolution 02/2004, as do other types of drugs, as explained in Chapter 4, Question 1.1. Companies must submit to CMED the request for price approval, after receiving marketing authorization from ANVISA and submit economic data on the product and propose a suggested price. Then CMED we will define the maximum price of the drug, using ERP and/or IRP.3. Describe the reimbursement process of orphan drugs in your country.
There is no specific reimbursement process for orphan drugs. They follow the procedure applicable for other drugs, as explained in Chapter 3, Question 3. Recently, Conitec issued a guideline for setting cost-effectiveness thresholds in health decisions. Scenarios were also discussed in which the threshold could have its values relaxed (including rare diseases, neglected endemic diseases, diseases in children, diseases affecting individuals at the end of life expectancy and high effectiveness in terms of QALY).4. Other than the clinical studies required, do the data and studies required for the pricing and reimbursement of orphan drugs differ from that of other drugs?
No, there is no difference.Also from this Market Access & Health Technology Assessment
13. Vaccines: Brazil
Join industry executives in staying informed on the market access and HTA process in Brazil.
1. What is the definition of a vaccine in your country?
As per ANVISA Resolution RDC 55/2010, vaccines are immunobiological drugs which contain one or more substances (antigens) that, when injected into an individual, stimulate the immune system to produce specific antibodies to combat these substances, protecting that individual against the disease caused by the agent that originated the antigen.2. What are the classifications of vaccines in your country?
Brazilian regulations do not categorise vaccines.3. Describe the pricing process for vaccines in your country (based on their classifications).
The pricing of vaccines follows the rules foreseen in CMED Resolution 02/2004, as do other types of drugs, as explained in Chapter 4, Question 1.1.4. Describe the reimbursement approval process for vaccines in your country (based on their classifications).
The reimbursement approval process for vaccines in Brazil is the same as for other drugs.5. Other than the clinical studies required, do the data and studies required for the pricing and reimbursement of vaccines differ from that of other drugs?
No, there is no difference.Also from this Market Access & Health Technology Assessment
14. HTA Dossiers in Brazilian Pharma
Part of PharmaBoardroom’s new country-focused series on market access and health technology assessment (HTA), this piece is focused on HTA Dossiers and Data requirements in the Brazilian pharma market today. Buy The Pharma Legal Handbook: Market Access & HTA – Brazil here for £359.
HTA DOSSIERS
- Have local authorities published recommendations surrounding value assessment dossiers? (If yes please add link)
- Have local Authorities published guidelines surrounding value assessment dossiers? (If yes please add link)
- Proposal Application Guideline;
- Methodological Guidelines - Budgetary Impact Analysis - Manual for the Brazilian Health System;
- Methodological Guidelines - Systematic Review and Meta-Analysis of Diagnostic Accuracy Studies.
- Have local authorities published official guidelines surrounding the submission of value assessment dossiers? (If yes please add a link)
- Describe the overall process of preparing and submitting a HTA dossier in your country.
- Describe the overall content of the HTA dossier in your country.
- Letter:
- The letter must be on the first page of the process and must contain: identification of the applicant; identification of the recipient (DGITS/SECTICS/MS); subject (name of technology and indication requested); place, date and signature.
- Applicant documentation
- For legal entities: articles of Incorporation of the company (notarized copy); and power of attorney of the applicant (in case the person responsible for signing the application is not included in the articles of incorporation).
- Application form
- The application form must be filled out and submitted using this link.
- Main document
- The document must contain: description of the disease/health condition related to the use of the technology; description of the technology; description of the scientific evidence of the technology compared to the one(s) already available at SUS (Systematic Review or Technical-Scientific Opinion); economic evaluation study from the perspective of SUS; budgetary impact analysis; bibliographic references; appendix (copy of package insert or instructions for use approved by ANVISA).
- Scientific studies
- Foreign language articles
- Foreign articles not in English or Spanish must contain a sworn translation into Portuguese.
- Economic and budget impact studies
- The studies must be available in .xlsm (Excel Macro Enabled Workbook File) or .trex (TreeAge) formats.
- Which are the questions to focus on when preparing a HTA dossier in your country?
- Which are the other strategic considerations to take into account when preparing a HTA dossier in your country?