The Pharma Legal Handbook: Chile





 The Pharma Legal Handbook: Chile answers essential questions about the legal and regulatory environment for pharmaceuticals in Chile. It is a must-have for any company operating in the country or looking to enter the market.
The Pharma Legal Handbook: Chile answers essential questions about the legal and regulatory environment for pharmaceuticals in Chile. It is a must-have for any company operating in the country or looking to enter the market.
Prepared in association with Carey, Chile’s largest law firm, it should answer any questions linked to regulation, pricing, clinical and preclinical trials, marketing, manufacturing, trademarks and patents, cannabinoid medicines, medical cannabis and opioid drugs.
If legal handbook content is updated, you will receive a free updated PDF for up to one year after purchase.
June 2019
1. Biosimilars & Biologics: Chile
1. Are biosimilar medicines considered the same as generic medicines in your country?
No. According to Technical Guideline No. 170, issued by the Ministry of Health on August 21, 2014 and approved by decree No. 945 of 2014 -and its amendments-, which contain the regulations for the sanitary registration of biosimilars (“T.G. No. 170”), a biosimilar is defined as a biotechnological medicine that has proven to be comparable in quality, safety and efficacy to the reference biotechnology product, based on its exhaustive characterization by means of comparability studies under equal conditions, consisting of comparative quality studies and comparative non-clinical and clinical studies.
Therefore, in no case can a biosimilar can be considered a generic product. This is confirmed in Technical Guideline No. 170 as it declares that “the normative framework for the authorization of generic medicines of chemical synthesis is well established as the demonstration of chemical identity and bioequivalence with the reference product, if it were the case, allow inferring the therapeutic equivalence of a product. However, this same criterion, as described, is not applicable to biosimilars, as these products are composed of complex entities, which are difficult to characterize by traditional methods”. In this regard, according to article 53 of Supreme Decree No. 03/2010 (“S.D. 03/10”), which sets forth our pharmaceutical product regulations, biological products cannot be submitted or filed under the simplified or generic pathway.
2. Are all biologic medicines, including biosimilar medicines, patentable in your country?
Biological medicines are patentable in accordance with our local Industrial Property regulations. However, in the case of biosimilars, it would only be possible to obtain patents associated with their manufacturing process, since the molecule itself would not meet the patentability requirements for obtaining a patent.
3. Is there a specific regulatory framework for the marketing authorization of biosimilar medicines in your country? If yes, what is the regulatory framework for the authorization of biosimilar medicines?
Yes, there is a regulatory framework for the approval of biosimilars in Chile.
The general rule is that biological products, including biotechnological products, should undergo a stand-alone procedure, submitting, thus, all full-fledged preclinical and clinical trials to accredit safety and efficacy.
Nevertheless, article 42, letter i) of S.D. No. 3/10, states that a Supreme Decree of the Ministry of Health shall set forth the Technical Guideline for the “abbreviation of clinical studies to evaluate the safety and efficacy of biotechnological products based upon the existence of another registered biotechnological product which utilizes the same active ingredient, unitary dosage, pharmaceutical form and route of administration”. This Technical Norm shall determine the active ingredients and their presentation for which this pathway is enabled (a biosimilar pathway). Our regulations emphasize that the applicant must submit “comparative studies” with the reference product.
In this regard and upon the previously cited provisions, the biosimilar pathway registration is only available for the active ingredients and their respective presentations included in T.G. No. 170 and the list of Reference Biotechnological Product (RBP).
T.G. No. 170 has been amended during the past few years so as to include more RBP (Decree No. 199/2016, Decree No. 50/2018 and Decree No. 30/2019) and it is expected to be further amended for including new RBP.
4. What kind of data package is needed to obtain approval for a biosimilar drug? Is this any different to the requirements for the original Biologics drug?
T.G. No. 170 is based upon the WHO Guidelines for similar biotherapeutic products and structures the biosimilar pathway upon a stepwise comparability process, head to head with the reference product (which is also specifically set within such guidance), for all stages of its development (characterization, non-clinical and clinical studies). It also includes provisions in connection to pharmacovigilance and extrapolation of indications.
Thus, the dossier package of a biosimilar must contain a full characterization dossier of a biological drug, plus comparability studies consisting on the characterization and evaluation of quality aspects, followed by head to head comparative non-clinical and clinical efficacy, safety and immunogenicity evaluation with the RBP.
5. What are the requirements for the choice of the reference comparator product?
T.G. No. 170 does not include a regulated procedure under which new active ingredients and reference biological products will get to be included within the same. Additionally it does not include a term within which new lists of active ingredients or reference biological products should be issued or amended. Therefore, the time and procedure for the inclusion of new products depends entirely on the Ministry of Health´s determination.
Indeed, T.G. No. 170 only states that the list of active ingredients and reference products will be updated in connection with the “state of the art” by resolutions issued by the Ministry of Health upon recommendations of the Public Health Institute (ISP). Based on such faculty, the Ministry decides when and how new products are included in the guideline, notwithstanding the ISP´s recommendations. Therefore, even though T.G. No. 170 entered into force at the beginning of the year 2014, only three updating resolutions have been issued (in 2016 through exempt decree No. 199, in 2018 through exempt decree No. 50 and in 2019, through exempt decree No. 30).
6. Can the comparator product be sourced from another regulatory jurisdiction? If yes, what are the data needed to support this approach?
As already explained, the biosimilar pathway is only available for the active ingredients and their respective presentations included in T.G. No. 170 and its amendments. Upon S.D. 03/10 (article 42, letter i) the reference product must be registered, but it does not make specific reference as if it is required to be registered in Chile.
Additionally, under T.G. No. 170 the reference biotechnological product must be (i) recognized as such by the national medicines sanitary authority; (ii) must have its own quality, safety and efficacy studies; (iii) the same RBP must be used throughout the comparability exercise; (iv) the RBP cannot be a biosimilar; and (v) the biosimilar candidate must have the same dosage form, active ingredient, unitary dose, indication(s), concentration and administration route as the RBP. Again, it does not state that it is required to be registered in Chile.
Indeed, upon the latest modification of T.N. No. 170 of April of 2018, the list includes a reference product which is not registered in Chile. Nevertheless, please consider that this is a very novel case, since it has only been included in April of 2018 and there has been no prior experience with these cases in Chile.
Finally, as mentioned in question 5 and according to T.G. No. 170, there is no official procedure for the inclusion of reference products in the list.
7. How are the prices of biosimilar medicines regulated? Is this any different from the requirements for the original Biologics drug?
Up-to-date, there is no price regulation for medicines in Chile.
8. What is the reimbursement policy for biosimilar medicine? Is this any different from the requirements for the original Biologic drug?
In Chile, there is no general reimbursement system or process. The owner of the product can determinate the sale price of a medicine, such as biologicals and biosimilars.
The Chilean healthcare system is primarily structured by a mandatory medical coverage which is required by law. This coverage is financed by health insurance contributions paid to the providers of healthcare insurance (FONASA or the ISAPREs) on a monthly basis by certain persons such as employees (in which case the relevant employers are legally bound to withhold the relevant amounts from employees’ monthly wages), independent workers, pensioners, etc. The law provides for a minimum medical coverage and the additional features depend on the health institution and the health plan chosen by each individual.
Additionally, there are two universal coverage programs, which are Explicit Guarantees in Health (GES plan) and the High Cost Treatment Financial Protection System (Ley Ricarte Soto), which cover specific treatments and medicines, some of which are, indeed, biotechnological products.
Isolated coverage has also been provided under the Extraordinary Auxiliary Fund administered of the MoH.
For further information, please see answers to queries No. 10 and 11 of Exhibit A.
9. Does biosimilar competition impact the reimbursement policy of the originator reference products?
No, please refer to question No. 8.
10. What is the legal framework for biosimilar medicines prescribing (clinical decision maker) and dispensing (pharmacy level, hospital or retail)? Is this any different to the requirements for the original Biologics drug?
The applicable legal framework is the same for any other pharmaceutical product.
Regarding prescription of medicines, this can be only performed by physicians (also dentists and midwives in their respective fields of competence and allowed medicines) based on the agreed decision of the attending physician.
On the other hand, the dispensing of medicines must be carried out by an establishment that must comply with all regulations applying to reception, storage, transport, distribution and dispensing of pharmaceutical products. Additionally, the professional who dispense the medicine must be in possession of the title of Pharmacist and be registered at the National Registry of Individual Health Providers. Products authorized for sale under prescription (Rx conditions) can only be sold in pharmacies and chemist’s stores (“Almacenes Farmacéuticos”). The professional must request the prescription before dispensing the medicine and should verify the compliance with prescription requirements.
11. Is the system considering physician-led switching and/ or pharmacy-level substitution (without involvement of the clinical decision maker)?
Please consider that T.G. N° 170 expressly states that “interchangeability or substitution” of biosimilars must be performed only under the decision of the treating physician, who will evaluate the risks and benefits, duly inform the patient and under a stepwise and controlled proceeding with strict medical supervision.
In this regard, our regulations do not permit the automatic substitution of a biosimilar product at the pharmacy level. This is a special provision for biosimilars, as under the Sanitary Code substitution by pharmacists of pharmaceutical bioequivalent products is permitted under the request of the patient, which is a provision applicable only to chemically synthesized products.
12. What are the post-authorisation requirements (including pharmacovigilance, risk management plans, post-approval studies) for biosimilar medicines? Is this any different to the requirements for the original Biologics drug?
The post-authorisation requirements for biosimilars are no different than biological and/or biotechnological drugs requirements.
Regarding the Pharmacovigilance of biotechnology and biosimilar products, the requirement of having Risk Management Plans (PMRs) is indicated and it is also indicated that said guideline indicates that it will be based according to what is stated in Title X of the Supreme Decree. No. 3/10 (S.D. No. 3/10) and Technical Guideline No. 140/12 (T.G. No. 140), identifying the biotechnological drug, its manufacturer, country of origin, INN, trademark and lot number.
13. Are there specific policies and requirements for labelling biosimilar medicines in the event of second medical use patents? Is this any different from the requirements for the original Biologic drug?
No, in Chile there are no specific policies for biosimilar labelling in the event of second medical use patents (e.g. carve outs or skinny labels).
14. Have there been any significant legal/judicial developments in relation to biosimilars in your country? (Including but not limited to IP, procurement, competition, misleading information campaign, access to reference comparator product)
Please take into account that, in Chile, there has been significant discussion in connection with the possibility of direct purchases for public procurement of products under which patients have initiated treatment, as such products could not be automatically substituted at the pharmacy level. The Chilean MoH, in consultation with the ISP, have indicated a treatment continuity policy for patients who have initiated treatment with a biotechnological product or a biosimilar enabling direct purchase for such products and not within a public bid. Such policy has been already implemented for several products within the High Cost Treatment Financial Protection System (Ley Ricarte Soto).
15. Are there proposals for reform or significant change to the legal, regulatory, procurement of biosimilars? If yes, when are they likely to come into force?
Please be aware that there is a bill of law currently in Congress (Bill of Law No. 9914-11) that contemplates interchangeability (in general) as one of its main topics.
In particular, this bill of law states that the Ministry of Health must incorporate in the National Drug Policy, an Interchangeability Strategy for Pharmaceutical Products and establish an Implementation Plan for it, which must be approved by resolution of the Minister of Health.
Also, it states that the Ministry of Health, by decree, must dictate, within six months following the date of publication of the resolution indicated in the preceding paragraph, a new Technical Guideline, at the proposal of the ISP, that determines the evidence to which must be submitted to pharmaceutical products to demonstrate their interchangeability. Said norm will determine the interchangeability tests according to the nature of the pharmaceutical products, among which will consider bioequivalence, good manufacturing practices, particle size and pharmacovigilance, among others.
In addition to the above, this bill of law also states that the ISP will determine the pharmaceutical products that are not interchangeable, which will be established in the sanitary registration (MA) of said products.
Thus, it may be possible that, in the future, the rule on interchangeability for biosimilars be modified as per the entering into effect of the provisions included in this bill of law.
Also from this Legal Handbook
2. Orphan Drugs & Rare Diseases: Chile
1. What is the definition of Rare Diseases in your country?
Currently, we don’t have a legal or regulatory definition for rare diseases in Chile. Indeed, neither our Sanitary Code nor Supreme Decree No. 03/2010 (S.D. No. 3/10), which sets forth our pharmaceutical products regulations contemplate a definition for rare diseases. Additionally, although mentioned in Law No. 20.850 which sets forth a High Cost Treatment Financial Protection System (commonly known as Ricarte Soto Law) rare diseases are not defined.
Nevertheless, Exempt Resolution No. 411 of February 2015 (Res. No. 411/2015) issued by the Public Health Institute (ISP) – which approves recommendations for the sanitary registration (Marketing Authorization – MA) of orphan drugs– does include a definition of rare disease as follows:
“Uncommon, minority, rare or orphan disease: one with prevalence less than 5 cases per 10,000 inhabitants”.
2. Does the designation of ‘Orphan Drug’ exist in your country? (Does it correspond with the definition of Rare Diseases?)
We have no specific legal or regulatory definition for orphan drug and S.D. No. 3/10 does not contemplate a definition or concept for the same.
Nevertheless, Res. No. 411/2015 does include a definition for orphan drug which is related or linked to the concept of rare disease set forth in that same resolution, as follows:
“Orphan pharmaceutical product or orphan drug: that medicine intended for the diagnosis, prevention or treatment of a rare disease or a condition whose aetiology has an equivalent frequency”.
In this regard, the ISP does not provide for a specific orphan drug designation process, but a pharmaceutical product could be considered as an orphan drug by meeting this concept.
3. What is the regulatory framework for the authorization of an Orphan Drug? (Is this regulatory framework based on Rare Disease status or can it alternatively be based on Orphan Drug foreign status?)
S.D. No. 3/10 does not contemplate a specific procedure for the authorization of an Orphan Drug.
Considering the above mentioned, the ISP on 2015 issued Res. No. 411/2015, including recommendations for obtaining the approval of an Orphan Drug. However, these recommendations indicate that the request must be made, among other requirements, through the abbreviated registration procedure –which, in case of an Orphan Drug should not last more than 4 months to obtain the registration–, providing evidence that the product is an orphan drug as certified by sanitary agencies such as the FDA or EMA and, additionally, requiring a prior resolution from the Ministry of Health (MoH) to that effect.
Unfortunately, to date there is no clarity on how to carry out this process before the Ministry of Health or how long this process will take in order for the MoH to issue such resolution. Therefore, Res. No. 411/2015 has not been as useful as intended, in the sense that it does not effectively guarantee a shorter term of approval of the registration under an abbreviated procedure.
Consequently, in case that an Orphan Drug registration is required, a good approach is to discuss it directly with the ISP, on a case by case basis.
4. Does your country have provisions for relaxed clinical trial/scientific evidence requirements in respect of Orphan Drugs as compared to other drugs?
Our regulations do not contemplate provisions for relaxed clinical trial/scientific evidence in respect of Orphan Drugs. Therefore, in principle, full clinical trial/scientific evidence must be submitted before ISP in order to obtain the sanitary registration of an Orphan Drug.
Nonetheless, Res. No. 411/2015, within its recommendations, indicates that as Orphan Drugs are unlikely to have phase III clinical trials, with a large number of patients, to evidence safety and efficacy as they are destined for the prevention, diagnosis or treatment of low prevalence or frequency diseases (both in their expression and agents). In this regard, the applicant may submit safety and efficacy clinical studies with a reduced number of patients, notwithstanding that the design and the risk/benefit profile will be evaluated and supported by the preclinical and clinical information available.
5. Is there an expedited pathway for Orphan Drugs?
Please see question No. 3 above.
6. Are foreign marketing authorizations recognized in your jurisdiction for Orphan Drugs? If yes, marketing authorizations from which countries are recognized?
No, any Orphan Drug intended to be commercialized in the Chilean territory must apply for a sanitary registration and must comply with the local requirements established in our regulations, regardless the fact that said product may have a registration in other jurisdiction.
7. Can Orphan Drugs be reimbursed? If so, is there a specific reimbursement procedure for Orphan Drugs?
In Chile, there is no general regulatory reimbursement system or process.
In this regard, orphan drugs will have coverage within the functioning of the Chilean Healthcare system.
The Chilean healthcare system is primarily structured by a mandatory medical coverage which is required by law. This coverage is financed by health insurance contributions paid to the providers of healthcare insurance (FONASA or the ISAPREs) on a monthly basis by certain persons such as employees (in which case the relevant employers are legally bound to withhold the relevant amounts from employees’ monthly wages), independent workers, pensioners, etc. The law provides for a minimum medical coverage and the additional features depend on the health institution and the health plan chosen by each individual.
Additionally, there are two universal coverage programs, which are Explicit Guarantees in Health (GES plan) and the High Cost Treatment Financial Protection System (provided by Law No. 2.8580, Ley Ricarte Soto), which cover specific treatments and medicines. Certain orphan drugs are included within the specific coverage provided by GES but main coverage for certain specific orphan drugs have been provided under Law No. 20.850.
Isolated coverage has also been provided under the Extraordinary Auxiliary Fund administered of the MoH.
For further information, please see answers to queries No. 10 and 11 of Exhibit A.
8. How are the prices of Orphan Drugs regulated?
Up-to-date, there is no price regulation for medicines in Chile. The owner of the product can determine the sale price of a medicine, such as Orphan Drugs.
9. In case of reference price based on a basket of countries, what countries are included?
Not applicable. See question No. 8.10. Have there been any significant legal/judicial developments in relation to Orphan Drugs in your country?
In Chile judicial activity for coverage of pharmaceutical products has not been historically high, but there have been increasing numbers of judicial cases brought for high cost medicines in the last years, including orphan drugs, mainly throughout constitutional protection actions.
11. Are there proposals for reform or significant change to the regulation of Orphan Drugs? If yes, when are they likely to come into force?
There have been a few bills of law which were subject to discussion in Congress for rare diseases and orphan drugs in Chile but with no active development.
Bill of law No. 7826-11 was submitted for discussion in August of 2011 and includes provisions on designation of orphan drugs, market exclusivity for orphan drugs and registration of patients and patient associations and protection of data privacy, among other matters. The bill has not had any movement since its submission.
Bill of Law No. 7643-11 was submitted for discussion in May of 2011 and it has similar provisions as Bill of Law No. 7826-11. Proceedings for this bill of law were recently reopened in July of 2019, but with no further discussion.
Finally, the bill of law for Medicines II (bill of law No 9914-11) which is in its final discussion phase in Congress, includes a provision allowing pharmacies, wholesalers and pharmaceutical laboratories to import raw material necessary for the treatment of rare or low prevalence diseases considered as orphan drugs by the local or international authorities, duly recognized as stringent regulatory authorities. Nevertheless, this provision has been recommended for its exclusion in the final discussion in Congress.
Also from this Legal Handbook
3. Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs: Chile
Cannabinoid Drugs
1. Are Cannabinoid Drugs authorized in your country?
So far, only pharmaceutical products containing an extract of flowers and leafs of cannabis sativa (Sativex solución para pulverización bucal) have been approved for sanitary registration (marketing authorization) by the Public Health Institute (ISP). We are also aware of an Import Authorization without sanitary registration granted to a company to import certain products containing cannabis.
2. What are the regulatory authorities with jurisdiction over Cannabinoid Drugs?
- Ministry of Health and ISP: regulate importation, registration, distribution, marketing and clinical studies for pharmaceutical products containing cannabis.
- Ministry of Agricultural and Agricultural and Livestock Service (SAG): Authorization of sowing, planting, cultivating or harvesting plant species of the genus cannabis.
3. Is there a specific regulatory framework for the authorization, pricing, and reimbursement of Cannabinoid Drugs?
Our local health regulations set the provisions for the authorization for the import, registration, distribution, marketing and clinical studies of Cannabinoid Drugs by the Ministry of Health and ISP. No regulations about pricing and reimbursement are included in the local health regulations.
4. Which are the cannabinoid drugs that have received market approval to date?
Please refer to question No. 1.5. Who can prescribe Cannabinoid Drugs?
A physician duly authorized to prescribe.6. Is there a list of doctors authorized to prescribe Cannabinoid Drugs?
No, there is no specific list in this regard.
Any physician duly authorized for the issuance of medical prescriptions may prescribe Cannabinoid Drugs.
7. What approvals or notifications are required to prescribe Cannabinoid Drugs?
The Cannabinoid Drug must have an approved sanitary registration (marketing authorization) granted by the Chilean Public Health Institute (“ISP”) in order to be commercialized in the country.
Cannabinoid Drug recognized in S.D. 404/85 may only be prescribed under retained prescription (receta retenida) with stock control.
8. Which organizations are authorized to sell/distribute Cannabinoid Drugs available?
In general, all entities which are authorized to sell/distribute controlled pharmaceutical products (authorized, pharmacies, wholesalers, among others)
9. Is there a list of retailers/distributors authorized to sell Cannabinoid Drugs?
No, there is no specific list in this regard.
10. Are there proposals for reform or significant change to the regulation of Cannabinoid Drugs?
Currently there are no bills or proposals to change regulations in this matter.
11. When are they likely to come into force?
Not applicable. See answer to question 10.Medicinal Cannabis
12. Is Medicinal Cannabis authorized in the country?
No. Chilean Law does not explicitly authorize the use of medicinal cannabis. Notwithstanding the above, there is a growing jurisprudence of the superior courts of justice, which has understood that if the cannabis is cultivated for medicinal use, the amount can be considered as “personal consumption” (which is authorized in some circumstances) and not for drug trafficking purposes (which can be criminally prosecuted).
13. What are the regulatory authorities with jurisdiction over Medicinal Cannabis?
N/A14. What is the regulatory framework for the authorization, pricing, and reimbursement of Medicinal Cannabis?
N/A15. How is the production and import of Medicinal Cannabis regulated and by which agencies/authorities?
N/A16. What approval or notifications are necessary to produce or import Medicinal Cannabis?
There is no applicable regulation for Medicinal Cannabis in this regard.
17. What is the regulatory framework for the marketing and distribution of Medicinal Cannabis?
N/A18. How can patients obtain Medicinal Cannabis?
N/A19. Who can prescribe Medicinal Cannabis?
N/A20. Is there a list of doctors authorized to prescribe Medicinal Cannabis?
N/A21. What approvals or notifications are required to prescribe Medicinal Cannabis?
N/A22. Where is Medicinal Cannabis available?
N/A23. Is there a list of retailers authorized to sell Medicinal Cannabis?
N/A24. Are there proposals for reform or significant change to the regulation of Medicinal Cannabis?
There is currently a bill in congress which intends to amend Law No. 20,000 with sanctions the illicit traffic of narcotics and stupefacient. Pursuant to this bill, medical prescription of cannabis will be deemed as an authorization to produce some products derived from cannabis, for medical treatment purposes. This bill provides that the use of medicinal cannabis under medical prescription cannot include “combustion”.
Opioid Drugs
25. Are Opioid Drugs authorized in your country?
Yes.26. What are the regulatory authorities with jurisdiction over Opioid Drugs?
- Ministry of Health and ISP: regulate importation, registration, distribution, marketing and clinical studies of opioid drugs.
- Ministry of Agricultural and Agricultural and Livestock Service (SAG): Authorization of sowing, planting, cultivating or harvesting plant species.
27. Is there a specific regulatory framework for the authorization, pricing, and reimbursement of Opioid Drugs?
Our local health regulations contemplate the authorization for the importation, registration, distribution, marketing and clinical studies of Opioid Drugs by the Ministry of Health and ISP.
Pricing is not regulated.
Reimbursement will depend on the private insurances of each patient.
28. Which are the Opioid drugs that have received market approval to date?
There are several Opiod drugs that have a sanitary registration (marketing authorization) in Chile containing the following active ingredients:
- Morphine
- Apomorphine
- Fentanyl
- Remifentanil
- Sufentanil
- Pethidine
- Oxycodone
- Hydrocodone
- Codeine
29. Who can prescribe Opioid Drugs?
A physician duly authorized for the issuance of medical prescriptions.
30. Is there a list of doctors authorized to prescribe Opioid Drugs?
No, there is no specific list in this regard.
Any physician duly authorized for the issuance of medical prescriptions may prescribe Opioid Drugs.
31. What approvals or notifications are required to prescribe Opioid Drugs?
The Opioid Drug must have an approved sanitary registration (marketing authorization) by ISP in order to be commercialized in the country. Opioids pertaining to Lists I or II as recognized in S.D. 404/85 may only be prescribed under official prescription (receta cheque) or retained prescription (receta retenida)
32. Which organizations are authorized to sell/distribute Opioid Drugs available?
In general, all entities which are authorized to sell/distribute controlled pharmaceutical products (authorized, pharmacies, wholesalers, among others)
33. Is there a list of retailers/distributors authorized to sell Opioid Drugs?
No, there is no specific list in this regard.34. Are there proposals for reform or significant change to the regulation of Opioid Drugs?
Currently there are no bills or proposals to change regulations in this matter.35. When are they likely to come into force?
Not applicable. Please refer to question No 34.Click the following links to read more legal articles from Chile:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
4. Regulatory Reform: Chile
1. Are there proposals for reform or significant change to the healthcare system?
A bill that will modify the Sanitary Code is currently under discussion. Main topics covered by this bill of law are: (i) prescription by INN; (ii) labeling; (iii) interchangeability of pharmaceutical products; (iv) inaccessibility; (v) medical reps; (v) value transfer regulations; (vi) price control mechanisms; (vii) non-voluntary licenses; (vii) advertising, among others.
2. When are they likely to come into force?
It is likely that this bill would be approved during 2021 (second semester).
Click the following links to read more legal articles from Chile:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
5. Patents & Trademarks: Chile
1. What are the basic requirements to obtain patent and trademark protection?
a. Patents:
Any invention that has novelty, inventiveness and industrial application may be subject to patentability.
In general terms, the basic requirements to obtain patent protection are the following:
- To complete a form with information regarding the applicant and the inventor, if applicable, or regarding the PCT Patent Application.
- A Spanish translation of the title, abstract, specification claims and drawings of the invention; if the application is filed under the PCT, it is necessary to submit the Performed Search Report and any modifications or declarations performed during the international phase.
- To file an original or certified copy of the assignment of the inventor/s to the applicant/s. In case of PCT applications, it is not necessary to file this document if the corresponding declarations where performed during the international phase.
- In case the applicant is foreign, it will be necessary to appoint a local representative and submit a hard copy of a Power of Attorney.
- In case a priority is cited, a certified copy must be filed, along with its translation into Spanish in a period of 90 days as of the filing of the application. In case of PCT applications, in which the priority has been properly submitted during the international phase, it will be required to file the same during the Chilean procedure only if requested by the Chilean Industrial Property Institute - Instituto Nacional de Propiedad Industrial (INAPI).
- Pay the corresponding official fees, publication fees and expert fees for the examination of the application.
b. Trademarks:
Any sign that is able to be graphically represented and capable to distinguish products and/or services may be subject to trademark protection.
To achieve trademark protection the interested party must file an application before INAPI and comply with the following basic requirements:
- Provide a graphic representation of the sign applied for registration, indicating its specific wording, figurative elements, and a description of the label or sound, if applicable.
- Indicate the products and/or services that the sign distinguishes.
- If the applicant is foreign, he must appoint a local representative and submit a hard copy of a Power of Attorney.
- Pay the corresponding official and publication fees.
2. What agencies or bodies regulate patents and trademarks?
Patent and trademark prosecution and cancellations are covered by the Instituto Nacional de Propiedad Industrial (INAPI) and by the Industrial Property Court. INAPI acts as a registrar entity and also as a first instance resolving authority. The Industrial Property Court is the second instance collegiate court in patent and trademark disputes, official rejections and cancellation actions. The Chilean Supreme Court may act on these proceedings in case a nullity remedy against a resolution issued by the Industrial Property Court is filed. Patent and Trademark infringement is under the jurisdiction of general civil courts.
3. What products, substances, and processes can be protected by patents or trademarks and what types cannot be protected?
a. Patents:
Any solution to a technical or art problem which originates an industrial task can be patented, provided it has novelty, inventiveness and industrial application.
Local regulations do not consider as an invention susceptible of patentability the following:
- Discoveries, scientific theories, and mathematical methods.
- Plants and animals, except microorganisms that comply with general conditions for patentability. Processes for the production of plants and animals which are essentially biological, cannot be patented, with exception of microbiological procedures.
- Easily verified and controlled economic, financial and commercial systems, methods, principles or economic plans and those referring to purely mental or intellectual activities or games.
- Surgical or therapeutic treatment methods for the human body or animals, as well as diagnostic methods applied to the human body or animals, except products used to implement one of these methods.
- The new use, change of form, dimensions, proportions or materials, objects or elements already known and employed for determined purposes. However, the new use of goods, objects or elements can be patent protected, provided that in addition to complying with the general patentability requirements, said new use solves a technical problem which did not have an equivalent solution, and that to obtain the new solution, changes in the objects or elements` dimensions, proportions or materials is required. The claimed new use must be proven by means of experimental evidence in the patent application.
- Parts of living beings as they are found in nature, natural biological procedures, biological material existing in nature or material that can be isolated, including genome or germoplasm. Nevertheless, procedures using one or more of the biological materials mentioned above and the products directly obtained by those procedures shall be patentable, provided they comply with the general patentability requirements, that the biological material is adequately described and that the industrial application of the same is explicitly outlined in the patent application.
- Inventions contrary to law, public order, national security, morality and good habits.
b. Trademarks:
Any sign that is subject of graphic representation and that is able to distinguish products, services and commercial or industrial establishments in the market may be registered as a trademark. Those signs can consist of words including names, letters, numbers, and figures such as images, graphics, symbols, color combinations, sounds, and any combinations of the same. Also, propaganda or publicity slogans can be registered, provided that they are added to a trademark registration.
In general terms, the following cannot be registered as a trademark:
- Any sign that is generic or descriptive of the products or services requested to registration.
- Any sign that can induce consumers to an error or confusion about any of the qualities or place of origin of the products or services that it distinguishes.
- The shape or color of the packaging of a product or a color itself.
- Scientific or technical denominations, Plant Varieties, Indication of Origin or Geographical Indications, and any common denomination recommended by the World Health Organization and those indicative of therapeutic action.
- Expressions against the morality and public order.
- The name or acronym of a state, international organization or public service, along with their symbols, such as their flag or emblems.
- Signs that reproduce or imitate official signs, seals or hallmarks indicating control or warranty, except if their registration is applied by the competent body.
- The name, pseudonym, portrait of a person unless such person or its legal representative gives its consent, or it has been more than fifty years since that person’s death and does not affect its honor.
- Signs identical or similar to trademarks registered in Chile.
- Signs identical or similar to trademarks registered aboard if these trademarks are famous and notorious.
- Any sign that may affect any third party’s legal right.
- Nontraditional signs such as tridimensional symbols, holograms, animated or multimedia material, smells, tastes and textures.
4. How can patents and trademarks be revoked?
a. Patents:
A patent may be revoked by means of a cancelation action filed before INAPI. The grounds for cancelling a patent according to the Chilean legislation may be any of the followings: the patent (i) was granted to a person who is not the true inventor or assignee, (ii) was granted over the basis of erroneous or evidently deficient examiner’s reports, and (iii) was granted in contravention of the rules of patentability (e.g. lack of inventive step or novelty).
The applicable statute of limitation is five years as of the date of the patent registration.
b. Trademarks:
A trademark may be revoked by means of a cancelation action filed before INAPI. The grounds for cancelling a trademark according to the Chilean legislation are any of the legal registration prohibitions (mentioned above). Currently, Chilean legislation does not recognize a cancelation action based on the non-use of a trademark. However, a new bill is being discussed by the Congress that includes use requirements for trademarks which might allow cancellation on these grounds.The applicable status of limitation is five years as of the date of the trademark registration. However, no status of limitation applies in case of trademarks obtained in bad faith.
5. Are foreign patents and trademarks recognized and under what circumstances?
a. Patents:
A patent must be registered in Chile to be recognized. However, if a patent has been previously requested abroad, the applicant has a one-year priority period to request the same in Chile.
Also, Patent Prosecution Highway (PPH), to speed up the national patent prosecution procedure is available due to international agreements with nine other jurisdictions, including Colombia, Mexico, Peru, Canada, Brazil and Japan, among others. The PPH conditions for each jurisdiction are regulated by specific guidelines.
PPH is available in both, Mottainai (in which the PPH request can be based on a favorable resolution obtained from a previous examination office) and PCT versions (in which the PPH can be requested based on a favorable result of the international phase examination).
b. Trademarks:
A trademark must be registered in Chile to be recognized. However, if a trademark has been previously requested abroad, the applicant has a six months priority period to request the same in Chile.
In addition, famous and notorious trademarks registered abroad can claim rights in Chile over an identical or similar trademark registered, or filed for registration, for the same products, services and commercial or industrial establishments, by filing a cancellation action or by opposing a pending application.
6. Are there any non-patent/trademark barriers to competition to protect medicines or devices?
Yes, Chile recognizes Regulatory Data Protection for Pharmaceutical pro-ducts for both chemcically synthesized products and biotechnological products. Articles 89 to 91 of Law No. 19.039 “Industrial Property Law” regulate the protection of undisclosed data regarding safety and efficacy of a pharmaceutical product submitted to the authority to obtain sanitary registrations or authorizations for a new chemical entity.
Such legislation entered into force in December of 2005, through the publication of its regulations contained in Supreme Decree No. 153 of the MOH, later replaced by Supreme Decree No. 107/08, which entered into force in December of 2010.
The data protected under our current legislation is undisclosed data of a new chemical entity. Article 2.1 of S.D. 107/08 defines this concept stating that “Test data and other information of undisclosed nature is the background information concerning the safety and efficacy of a pharmaceutical product comprising a new chemical entity, such information consisting in complete studies with sufficiently developed information on the basis of clinical and pre-clinical trials”. This will cover preclinical and clinical study data, whether in phase I, II or III.
RDP lasts 5 years since the sanitary registration of the pharmaceutical product containing the new chemical entity.
In this regard, the ISP shall:
- Maintain confidential all information with RDP.
- Not use the protected data for granting a sanitary registration, unless authorized by the holder of the protected data.
Furthermore, article 53 (b) of S.D. 03/2010, states that the simplified procedure (which corresponds to the generic pathway) is not available when “The pharmaceutical product for which registration is sought comprises the same active ingredient as another registered product, when the information regarding such product is protected under the provisions of section 2, title VIII of law No. 19,039 , granted in accordance to the specific regulations governing the matter, or when it is based in the data having such protection”. Article 53.b thus provides that a generic application for a generic pharmaceutical product may not be filed during Regulatory Data Protection period.
7. Are there restrictions on the types of medicines or devices that can be granted patent and trademark protection?
Yes. Dosages, Surgical or therapeutic treatment methods for the human body or animals, including administration routes and administration regimes, as well as diagnostic methods applied to the human body or animals, are not patentable in Chile.
Second medical use is accepted in Chile but only under a very strict evaluation criterion. In this sense, claims must be drafted using the Swiss format, and the same must be well supported with experimental evidence in the description.
8. Must a patent or trademark license agreement with a foreign licensor be approved or accepted by any government or regulatory body?
No, license agreements are not subject to the approval of any entity, nor is their registration required before the INAPI or any other official body. However, for a license to be enforceable against third parties it must be duly recorded before the INAPI.
Click the following links to read more legal articles from Chile:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
6. Product Liability: Chile
1. What types of liability are recognized in your jurisdiction?
In our jurisdiction, the liability system is mainly based on negligence.
Regarding defective products, since there are no special liability rules, general rules of liability will be applicable. General rules are set forth in Chilean Civil Code regarding tort liability and Criminal Code, as well as in Consumer Protection Act (CPA) and the Sanitary Code.
Penalties and sanctions for non-compliance of sanitary laws and regulations may be imposed by the ISP prior carrying out the respective administrative sanctioning procedure (sumario sanitario). The penalties which it may impose upon finding a sanitary infringement include fines ranging from USD 0.1 to USD 70,000. In case or repeated infringements, previously imposed fines can be duplicated.
Additionally, the ISP can shut down the location or building where the infringement is committed, prohibit its operation, revoke the permits or authorizations granted, suspend distribution of products, among other sanctions included in section 174 of the Sanitary Code.
Regarding product liability, the Consumer Protection Act (CPA) recognizes two type of actions in order for the supplier to be held liable: (i) noncompliance and (ii) civil actions. Noncompliance or law infringement usually results in the application of fines and other measures (e.g. recalls, advertisement amendment, etc.); and civil liability is originated from damages caused to consumers.
General rule is set forth in article 23 of the CPA: suppliers are liable when they neglectfully cause damages to consumers in the sale of a product with flaws or deficiencies in its quality, quantity, identity, substance, origin, safety, weight or measure. This infringement has two consequences: (i) as a law infringement, it can be sanctioned with a fine of up to 300 UTM (US$ 21,700 approx.); and (ii) as civil liability, consumers are entitled to pursue damages from the supplier for moral and material damages. The CPA expressly establishes a basic right for consumers to be adequately compensated in a timely manner for all material and moral damages caused by a supplier that breaches its obligations.
In line with product liability, CPA sets forth several rules applicable to hazardous and risk services or products, e.g. obligation to provide specific information to consumers and the general public. Moreover, if an unforeseen risk or danger in a product arises after it was introduced in the market, the supplier must inform the competent authorities (i.e. the aforementioned health organisms) and adopt the necessary preemptive and/or corrective measures. The breach of this duty can be sanctioned with a fine of up to 50 UTM (US$ 3,600 approx.).
Article 49 of the CPA establishes that any breach of the obligations mentioned in the preceding paragraphs will subject the liable supplier to the noncompliance sanctions, to the respective compensation for all damages caused and the applicable criminal penalties if such events constitute a crime or felony.
Moreover, please bear in mind that Sanitary Code includes an additional set of rules for defective sanitary products in articles 111 H to 111 N:
- Defective sanitary product is the one that is not safe enough, having in mind all the circumstances related to the product and, especially, its presentation and its reasonably foreseeable use. Furthermore, a product is deemed as “defective” if it does not offer the same safety normally found in the rest items of the same series.
- A product cannot be considered defective by the only fact that it was subsequently released to market in an improved manner.
- Every damage caused using a defective sanitary product will result in civil and criminal liability, as applicable.
- Authorizations holders, manufacturers and importers will be responsible for the damages, as applicable. The ones responsible for the damage will be jointly and severally liable to the injured parties. Anyone who responds to the injured party shall have the right to seek for reimbursement from the remaining responsible, pursuant to their participation in the creation of the damage.
- The damaged party who seeks for indemnification will have to prove the defect, the harm caused, and the causal link between both.
- In clinical trials, once proven, the damage will be presumed to have been produced on the investigation.
- The defendant will not be exempted from liability arguing that the damage caused by a defective sanitary product arises from events or circumstances that were not foreseen according to the state of the scientific or technical knowledge existing at the moment of use.
- The statute of limitations is five years counted from the date of appearance of the damage, either due to the defect of the product or the damage caused by such defect. The statute of limitation for damages caused with the occasion of a clinical trial is ten years.
- The manufacturers and importers of sanitary products must take out insurance, an endorsement or equivalent financial guarantee to respond for health damages that arise from safety problems.
2. How do these types of liabilities apply to the manufacturers of medicines and devices?
Regarding sanitary penalties as stated above, under our regulations the owner of an authorized facility shall be jointly responsible together with its technical director for the correct distribution and sale, at any title, of the products imported or commercialized by such facility or for the advertising and information provided for such products. In any case, the responsibilities affecting the Technical Director, Head of Production, Head of Quality Control and Head of Quality Assurance of authorized facilities shall always affect the responsibility of the owner of the facility.
Regarding responsibility under the CPA, and pursuant to its broad definition of supplier, manufacturers are considered as such; hence, their general obligations and liabilities are applicable.
Further, there are specific cases in which CPA expressly sets forth the manufacturer’s liability. In case a hazard or toxicity that risks the health or safety of consumers is determined or established by the competent authority or court, the manufacturer will be jointly liable along with the importer and first distributor. In turn, the supplier will have to proceed at its own expense to change all the products to others that are safe, with equivalent price and utility. If this is not possible, the supplier will have to reimburse consumers with all paid prices.
Please refer to Question 1 above, too.
3. Does potential liability extend to the manufacturer only or could claims extend to corporate executives, employees, and representatives?
As previously indicated, sanitary sanctions can also reach specific employees of the manufacturer of a pharmaceutical product. Specifically our regulations set forth personal responsibilities and liability for the Technical Director, Head of Production, Head of Quality Control and Head of Quality Assurance, which will also always reach the owner of the facility.
On the other hand, general liability rules and CPA responsibility described in answers 1 and 2 are applicable to the vendor, distributor and/or the manufacturer when applicable, and it does not extend to corporate, executives, employees and/or representatives, unless a criminal offence can be established pursuant to the Criminal Code or personal liability be generated under tort regulations.
Please refer to Question 1 above, too.
4. How can a liability claim be brought?
Liabilities can be pursued by authorities and affected consumers. The health authorities can officiate and initiate an administrative sanctioning procedure (sumario sanitario) against the manufacturer ex officio or through a complaint filed by an end-user.
From the consumer protection standpoint, the authority in charge is the National Consumer Service (SERNAC). Any affected consumer for a CPA breach can occur before SERNAC, in order to initiate a voluntary individual mediation process. SERNAC can perform collective mediations as well. However, these procedures are not mandatory, and the authority does not have the faculties to impose sanctions directly.
In addition, and in order to ensure compliance with the CPA, SERNAC is entitled to request information and/or inspect suppliers. In case of a law infringement, any affected consumer or the authority may file a judicial complaint based on an individual interest. Class actions can be filed by SERNAC, a Consumer’s Association, or a group of fifty or more consumers.
Finally, a civil or criminal lawsuit regarding damages may also be brought pursuant to the general rules.
5. What defenses are available?
One of the main defenses for suppliers –manufacturers, distributors, importers, vendors–involved in the chain of production of pharmaceutical and medicinal products, is the complete compliance with all the legal and regulatory requirements for the manufacture, import and distribution of said products, and keeping record of such compliance. The aforementioned would be useful in case of a potential trial, in order to argue that the contested products comply with the applicable regulations and sectorial requirements.
Click the following links to read more legal articles from Chile:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
7. Traditional Medicines and OTC Products: Chile
1. What are the regulatory requirements for traditional, herbal, complementary, or alternative medicines and devices?
In Chile, Traditional Herbal Medicines shall be understood as those constituted by plants or parts of plants, fresh or dried, whole or crushed, packaged and labeled by hand and labeled with the commonly used name in the field of Chilean cultural traditions, which have been recognized in the respective technical rule approved by Supreme Decree of the Ministry of Health. These medicines must:
- Be in a list contained in a technical rule approved by Supreme Decree of the Ministry, issued in use of their legal technical regulatory powers, which will indicate the denomination, therapeutic properties and uses of each of them, should be used as auxiliaries symptomatic.
- Be hand-packed as isolated plant species, not mixed.
- Include in their labels only those properties recognized in the aforementioned decree.
Resolution N° 548/2009 issued by the Ministry of Health approved the list of Traditional Herbal Medicines allowed in the country and their medical properties.
Our regulation does not include a definition for complementary or alternative medicines/devices; however, any product claiming a therapeutic property shall be considered a pharmaceutical product and therefore, it must be subject to regulation regarding pharmaceutical products.
2. Can these traditional, herbal, complementary, or alternative products be advertised directly to the public?
Traditional herbal medicines are considered pharmaceutical products as per article 7 of S.D. No. 3/2010; therefore, they are subject to regulation regarding pharmaceutical products. See Chapter about Marketing, Manufacturing, Packaging and Labeling Advertising – question 20.
3. What health, advertising, and marketing claims may be made for traditional, herbal, complementary, or alternative products?
For Traditional Herbal Medicines, it is only possible to display those properties and traditional uses as recognized by Resolution N° 548/2009.
4. What are the regulatory requirements for over-the-counter (non-prescription) medications?
The regulatory requirements for the registration of OTC products are the same that apply for prescription products (See Chapter 1 – question 3B), with the following considerations:
- If they are well known products, they can be registered under simplified procedure (no safety and efficacy studies are required);
- The advertising of OTC medicines is allowed but it must be previously approved by the ISP;
- The sale of OTC products can be performed on shelves, dispensers or other similar devices, which can be used solely for the exhibition and sale of OTC products in a pharmacy having a previous approval from ISP to said purpose.
5. Are there any limitations on locations or channels through which OTC products may be sold?
Please refer to Chapter: Marketing, Manufacturing, Packaging & Labeling, Advertising, question 18.6. What health, advertising, and marketing claims may be made for OTC products?
All claims must be in accordance with the approved registration information (indication, mode of use, etc.) and marketing claims and advertising must be previously approved by ISP.
7. Can OTC products be marketed or advertised directly to the public?
Yes. Please refer to Chapter: Marketing, Manufacturing, Packaging & Labeling, Advertising, question 20.8. What is the mechanism by which a prescription-only product can be converted to an OTC product?
To switch the sale condition of a pharmaceutical product from “under prescription” to “OTC”, the Ministry of Health issued Resolution No. 1133 of July 2001 (and its modification, Resolution No. 779 of November 2005), approving the Technical Rule No. 58, which establishes the criteria for OTC switch.
The criteria to perform this change correspond to safety criteria (wide therapeutic margin, use of at least 10 years in the country, no severe side effects, among others), criteria that allow determining the condition of direct sale (use to prevent symptoms of easy recognition, not to be used parenterally) and criteria for the information of the user (information to the patient must be complete, there may be specific requirements for its labeling, etc.).
9. What are the requirements for the importation of either traditional medicines or OTC products?
Same as for prescription products, that is, they must have a sanitary registration approved by ISP in order to be imported into the national territory.
Click the following links to read more legal articles from Chile:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
8. Marketing, Manufacturing, Packaging & Labeling, Advertising: Chile
1. What is the authorization process for the marketing of new drugs, biologics, medical devices, over-the-counter medications, and other medicinal products?
Please refer to Chapter 1: Regulatory, Pricing, and Reimbursement Overview, question 3.2. What is the authorization process for the marketing of generic versions of these products?
Please refer to Chapter 1: Regulatory, Pricing, and Reimbursement Overview, question 3.3. What are the typical fees for marketing approval?
Please refer to Chapter 1: Regulatory, Pricing, and Reimbursement Overview, question 4.4. What is the period of authorization and the renewal process?
The period of authorization for pharmaceutical products is five (5) years. The renewal process follows the same pathway as the marketing authorization but with minor requisites upon ISP request.
For imported products, our regulations require that the application for renewal be submitted together with the Certificate of Pharmaceutical Product, Registration Certification, Sanitary Authorization Certification or the Official certification recommended by the WHO, issued by the sanitary authority of the country of origin and which evidences that the manufacturing or storage facility, as applicable, meets the conditions required by the laws of such country, that the product has sanitary registration and indicating the complete authorized formula and if the sale of the product is subject to any restrictive sanitary regime or control, if applicable.
As per article 55 of S.D. 03/2010, the following conditions must be met in order to obtain the renewal of the sanitary registration of a pharmaceutical product:
- Payment of the relevant governmental fee.
- Compliance with observations made for the suspension of the sanitary registration within the timeframe granted for such purpose, if applicable.
- Non-existence of unpaid fines or compliance with other sanitary measures or sanctions imposed by the ISP in relation to the sanitary registration intended for renewal, if applicable.
5. What are the requirements, if any, for post-approval pharmacovigilance?
Resolution No. 381/2012 issued by the Ministry of Health approved the General Technical Standard on the National Pharmacovigilance System of Pharmaceutical Products for Human Use (hereinafter Technical Standard No. 140) that establishes those who are entitled to participate in pharmacovigilance activities and the actions that must be carried out for each of the participants in the national pharmacovigilance system.
For sanitary registration holders, post-approval pharmacovigilance is regulated by Resolution No. 108/2013, which establishes the content and frequency of Adverse Events and Adverse Reactions reports to be reported to ISP.
The holder of a sanitary registration must submit all reports associated with the safety of its products as well as alerts or health problems with their product(s) in other countries to the ISP through a platform.
The ISP will request, in qualified cases and through a well-founded resolution, a Risk Management Plan (RMP) for products corresponding to molecules first introduced in the market, biotechnological products and similar products to innovators that already have an RPM.
The RPMs are requested through the resolution of the ISP that approves the product sanitary registration (resolution of approval), for which a period of 60 calendar days, counted from the date of the presentation, is granted.
In relation to Periodic Safety Reports (PSURs), in accordance with Technical Standard No. 140, these should be required for innovator products, in order to strengthen the safety data thereof.
The frequency of the PSURs will be determined according to the date of the first authorization granted to any pharmaceutical company in any country according to the following scheme:
- During the first two years of commercialization, the report must be submitted every six months at the most;
- During the next three years; annually as a maximum; and subsequently;
- Every five years at most.
6. Are foreign marketing authorizations recognized?
No. It is mandatory to obtain a local sanitary registration to sell and distribute the product in Chile.7. Are parallel imports of medicines or devices allowed?
From an IP perspective Chilean Industrial Property Law N° 19.039 recognizes the exhaustion of industrial property rights related to trademarks and patents.
Regarding trademarks, article 19 bis E states that the right conferred by the trademark registration does not enable the right holder to prohibit its use by third parties related to the products legitimally commercialized in any country with such trademark by the holder or with its express consent. In the case of patents, article 49 states that patent for an invention shall not confer the right to prevent third parties from marketing the product protected by the patent, which they may have legitimately acquired after said product has been legally introduced into the market of any country, by the owner of the right or by a third party, with the owner’s consent.
Nonetheless, the import, distribution or sale in Chile of any pharmaceutical product requires a prior sanitary registration or authorization granted by the ISP.
8. What are the restrictions on marketing practices such as gifts, sponsorships, consultancy agreements, travel and entertainment, or other incentives for healthcare organizations and individual medical practitioners?
Regarding promotional activities, S.D. 03/2010 allows to provide information on pharmaceutical products to healthcare professionals who can prescribe or pharmacists who can dispense them. Such information must be on label and in accordance to the information approved for the product within the sanitary registration (except for information on off label indications or dosages where it is disclosed to the professional that they have not been approved in Chile to be used under his/her responsibility), complete, truthful and balanced.
Under S.D. 03/2010 it is forbidden to give incentives in any shape or form such as money, goods, services or others to healthcare professionals responsible for prescribing or dispensing medicines, or to individuals selling them, to encourage the prescription, dispensation or sale of a product over another. Additionally, the sale of pharmaceutical products may not be encouraged by giving incentives in any shape or form to pharmacy staff.
On the other hand, under the Sanitary Code, it is forbidden to provide economic incentives of any nature which may induce any person to prefer the use, prescription, dispensation, sale or administration of one or more pharmaceutical products. Moreover, the Ministry of Health may include in this prohibition some elements of medical use.
An incentive is understood to be any payment, gift, service or economic benefit granted or given to the above mentioned persons, by pharmaceutical laboratories, wholesalers, importers, distributors, pharmaceutical establishments in general or by their representatives, in general, by any person or entity that has an interest in preferring the use of one or more products or devices.
Furthermore, certain industry codes include further restrictions and regulations regarding marketing practices such as gifts, gimmicks, sponsorships, consultancy agreements, travel and entertainment.
9. How is the manufacturing of medicines and devices regulated and by which agencies?
The ISP is the entity in responsible for the oversight of manufacturing of medicines and devices in Chile.
The manufacturing process of pharmaceutical products is regulated by S.D. No. 03/2010. Article 108 of S.D. No. 03/2010, states that every production laboratory must comply with the rules contained on the Good Manufacturing Practices. Good Manufacturing Practices regulations are approved in Chile by Technical Rule N° 127, issued by Decree No. 28 in 2012 of the MOH and later modified by Decree N° 159 of the MOH in 2013.
According to article 112 and 167 of S.D. N° 03/2010, the ISP may inspect any facility of a production laboratory to verify its functioning conditions in connection to the regulations and Good Manufacturing Practice.
Additionally, every holder of a sanitary registration shall evidence that the manufacturing of the pharmaceutical product, whether local or foreign, meets the concept of quality assurance in the sense of accrediting that manufacturing and analysis methods are validated and lead to obtaining products in compliance with quality requirements, set forth in the sanitary registration in accordance to Good Manufacturing Practices.
The manufacturing of pharmaceutical products is inspected by ISP based on the content of Resolution N° 1290 of April 2016, which approves the “Guideline for Inspection of Public Health Institute”.
For medical devices, the regulations are established in Supreme Decree N° 825/98.
10. Are local manufacturing requirements compatible with Good Manufacturing Practices (GMPs) as defined by the U.S. Food & Drug Administration and/or the European Medicines Agency?
Yes. Good Manufacturing Practices regulations are issued and based upon the international recommendations set forth by the WHO.
11. What is the inspection regime for manufacturing facilities?
The inspection regime is regulated in Technical Rule N° 127.
The areas regulated are, in general, the following:
- Quality assurance.
- Good Manufacturing Practices.
- Sanitization and Hygiene.
- Qualification and Validation.
- Complaints.
- Recall of products.
- Manufacture and analysis by contract.
- Self-inspection and quality audits.
- Staff.
- Training.
- Staff hygiene.
- Facilities.
- Equipment.
- Materials.
- Documentation.
- Good practices in production.
- Good practices in quality control.
There is also a long standing guideline for inspection of GMP, issued by the ISP in 1997.
12. Are manufacturing facilities open for inspection by foreign inspectors or third-party inspectors as authorized by the FDA/EMA?
Our regulation does not cover or mention the possibility for manufacturing facilities to be open for inspection by foreign inspectors or third-party inspectors as authorized by the FDA/EMA. Nonetheless, manufacturing facilities are inspected and certified by foreign authorities such as EMA and Anvisa. There are also cooperation agreements signed by the ISP with FDA and with the Pacific Alliance countries for collaboration in sanitary registration and GMP certification of chemically synthesized products.
13. What are the requirements for storage, packaging, and handling of medicines and devices and their constituent components?
Technical Rule 147 of Good Storage and Distribution Practices for warehouses and deposits of pharmaceutical products for human use, approved by Decree N° 57 issued in February of 2013 by the MOH, sets forth the requirements for storage, packaging, and handling of medicines.
In general, Technical Rule N°147 sets forth requirements for storage of pharmaceutical products in matter such as (i) Facilities and installations; (ii) Requirements for storage; (iii) Returned products; (iv) Dispatch and transport; and (v) Recall.
Additionally, it regulates the distribution of pharmaceutical products in matters concerning (i) Organization; (ii) Staff; (iii) Quality system; (iv) Storage and warehouse; (v) Vehicles and equipment; (vi) Transport containers and labeling of containers; (vii) Dispatch and reception; (viii) Transport and products in transit; (ix) Documentation; (x) Complaints; (xi) Recall from the market; (xii) Returned products; (xiii) Falsified pharmaceutical products; (xiv) Import; (xv) Contract services; (xvi) Self-inspection.
Exempt Resolution 1422 issued by the ISP in 2014 sets forth the amended Guidelines for Inspection of Good Storage and Distribution Practices.
14. What information must be included in medicine and device labeling?
Pharmaceutical products in General:
Pursuant to the requirements set forth in S.D. 03/2010, the label of the secondary packaging of a pharmaceutical product shall be in Spanish, in clearly visible letters, and is required to include at least the following information without any advertising or promotional texts.
- Name of the pharmaceutical drug.
- Pharmaceutical form and dosage unit in the case of mono-drugs.
- Unconventional release pharmaceutical forms shall be indicated as such on the packaging material, as stated in the corresponding registration.
- Number of dosage units.
- Formula composition: quantitative and qualitative list of active ingredients and excipients.
- Name and address of the holder and the manufacturing laboratory, packer or importer, as appropriate.
- Route of administration.
- Approved retail conditions indicated with the appropriate abbreviation or full text.
- Expiry date. Furthermore, the included or recommended solvent shall also be required to be indicated in the case of extemporaneous preparations, as well as the effectiveness period after reconstitution, if applicable.
- Registration number given by the Institute, preceded by the following acronym “Reg. I.S.P.” (Institute of Public Health registration).
- Product code. Imported finished products shall keep the original code.
- Storage and conservation conditions.
- Incorporation of the caption, “More information on www.ispch.cl”, and others as set forth in article 87, if applicable.
- Any other information specially and additionally required hereunder or deemed necessary by the Institute upon registration or later.
Secondary packaging of bioequivalent product must include the “bioequivalent isologo”.
The primary packaging shall be required to include at least the information described in items 1, 2, 7, 9, 10 and 11 of the indicated above.
The ISP issued in 2014 and instructive guideline for labelling of pharmaceutical products.
Medical Devices:
Regulated medical devices must include in their labels the specific information set forth in the Chilean Official Norms (NCh) as made applicable in their respective implementing decrees.
In general, they must include, in Spanish the following minimum information:
- Name of the product.
- Intended use
- Name and address of the manufacturer.
- Name and address of the distributor in Chile.
- Expiration date.
- Sterilization method, if the product is sterile.
- Lot number.
- Storage conditions.
- Quantity of product in package.
- Indication if product is of single use, if applicable
Non-regulated devices, are not subject to such provisions, but general consumer protection rules shall apply, under Law No. 19,496 Consumer Protection Act (CPA). In this regard, please note that, according to the CPA, basic commercial information of the products, its identification, instructions of use, warranty and promotion/advertising must be made in Spanish language, in local currency (Chilean pesos), and using the official weight and measurement system (metric system).
Regarding the specific labeling of the product, it is necessary to include the identification of the products in Spanish (although it is acceptable for such information to also be provided in a different language, in addition to Spanish). Moreover, any other information set forth in the labels and/or use guides/user manuals must also be included in Spanish.
Please note that all the information mentioned above can be added to the device by means of a label sticker.
15. What additional information may be included in labeling and packaging?
Pharmaceutical drugs packed as medical samples shall be required to bear the caption “MEDICAL SAMPLE NOT FOR SALE” (in Spanish) in both the primary and the secondary packaging in indelible ink, in a clear and visible manner.
The labels shall be required to be printed or adhered to the outer surface of the package without being in contact with its contents. Arial or other similar straight fonts shall be used for labelling purposes, in a minimum size of 6.
The graphic label of imported finished pharmaceutical products may exceptionally contain texts in other languages in addition to Spanish, provided that the text authorized by the Institute is not modified.
In addition to the information described in question 14 above, pharmaceutical products intended for direct sale shall be required to include the following details:
- Regular dosing instructions for each indication as authorized in the corresponding registration.
- Any warnings as deemed necessary for safe and effective use of the product, indicating contraindications, interactions and adverse reactions if applicable as determined by the Institute upon granting the sanitary registration.
- These products may be presented in dispensers provided that each blister or strip is placed within an insert or other unit disclosing the full text of the caption approved for the secondary packaging and the patient information leaflet, if not enclosed.
Also, the holders of sanitary registrations may include in the label of pharmaceutical products, the markings or language related to industrial property rights (trademarks and patents) as required by law to exert the rights therein conferred.
16. What items may not be included in labeling and packaging?
S.D. 03/2010 expressly states that advertising or promotional texts may not be included in the label of pharmaceutical products.
According to the general rules under CPA, the information included in the products, labels, packaging, advertising of goods and services must be capable of verification and shall not contain expressions that mislead or deceive the consumer.
17. What are the restrictions and requirements for the marketing and advertising of medicines and devices?
Pharmaceutical prescription drugs -.i.e. with condition of sale under simple, retained or official prescriptions- cannot be advertised nor be subject to other activities to make the product known. Prescription drugs can, however be informed and promoted directly to the prescribing physician or the dispensing pharmacist.
Pharmaceutical products which are authorized as direct sale pharmaceutical products (OTC) can be advertised to the public. Nevertheless, the advertising must be previously approved by the ISP.
The advertising may only reproduce, totally or partially, the content of the patient information leaflet and labelling, as approved in the respective sanitary registration. Also, it may only refer to the therapeutic recommendations approved by the ISP in the sanitary registration and, in no case, may include titles, figures, indications, effects or other information not in conformity with the sanitary registration.
For devices, there is no specific regulation on advertising and marketing of medical devices. Therefore, general rules under CPA must be taken into consideration.
18. Where can medicines and devices be sold or delivered? Can medicines and devices be sold or delivered via post?
As per the provisions set forth in the Sanitary Code, the commercialization of pharmaceutical products is limited only to pharmacies and other authorized facilities duly authorized by ISP.
In connection with the display of pharmaceutical products in pharmacies, pursuant to the Sanitary Code it is possible to deploy shelves, dispensers or other similar devices, which can be used solely for the exhibition and sale of OTC products. It would be possible to display the product itself, the empty package, as well as other devices with images or mock-ups that represent it. To this aim, pharmacies must have an exclusive and defined area inside the facility which allows customers to have immediate access to it, duly authorized by the ISP.
With regards to the possibility of delivering pharmaceutical products via post, please consider that, as stated above, since the commercialization of pharmaceutical products is limited only to pharmacies and other authorized facilities, it follows that the dispatch of products must necessarily be performed exclusively from such locations. Furthermore, these facilities require a “brick-and-mortar” facility as the authority, for its installation, will require the location of the facility and the legal documents that accredit the property over of the same.
The shipping of the products will be mediated by means of an specific delivery service, such as postal offices.
In addition, the dispatch of pharmaceutical products will require the approval of the technical director of the pharmacy or the respective authorized facility. Moreover, in accordance to article 129 A of the Sanitary Code, one of the responsibilities of such technical director at the pharmacy is to carry out or supervise the proper dispensing of pharmaceutical products, in accordance with the terms set forth in the prescription, to inform personally and to promote their rational use, absolving the queries made by users.
For devices, there is no specific regulation or restriction in this regard.
19. What are the restrictions and requirements for electronic marketing and advertising via email, by Internet, social media, and other channels?
For pharmaceutical products, this kind of advertising is allowed solely for OTC pharmaceutical products subject to prior approval granted by the ISP.
If the ISP approves this kind of advertising, then the CPA rules shall be taken into account: any commercial communication addressed to consumers by email must clearly indicate: (i) the identity of the sender, (ii) the subject, and (iii) a valid form to request the suspension of these communications (i.e. unsubscribe).
Furthermore, Article 28 B of the CPA provides that commercial communications must grant an “opt-out” mechanism, as recipients of commercial communications by electronic means shall always have the right to request that any such communications cease, through easily accessible means available in Spanish. Thus, if a consumer chooses to opt-out the supplier must suspend any further communication.
For devices, there is no specific regulation or restriction in this regard and the general rules of the CPA shall apply.
20. May medicines and devices be advertised or sold directly to consumers?
For pharmaceutical products it will depend on their approved sale condition (prescription medicines or OTC medicines). In this regard, only OTC medicine can be advertised and sold directly to consumers only in pharmacies or other duly authorized facilities. However, all advertising of OTC medicines must be previously approved by the ISP.
For devices, there is no specific regulation on advertising and marketing of medical devices. Therefore, general rules of the CPA shall apply.
21. How is compliance monitored?
Through inspections carried out by the ISP as a result of its control plan or inspections from ISP originated by third-party complaints.
22. What are the potential penalties for noncompliance?
Please refer to Chapter about Regulatory, Pricing and Reimbursement, Question No. 9. In cases of repeated infringements fines can be duplicated.
Additionally, the ISP can shut down the location or facility where the infringement is committed, prohibit its operation, revoke the permits or authorizations granted, suspend distribution of products, among other measures (Section 174 of the Sanitary Code).
In the case of advertising please consider that the sanitary registration of a product may be suspended if the information of advertising or medical promotion is found as non-corresponding to the information approved for the sanitary registration of the product. Any non-conforming advertising or information to the professionals may be also suspended by the ISP who can issue and order for the immediate ceasing or withdrawal of the advertising.
Finally, additional to the registration holders, any individual or entity participating in the divulgement of unauthorized publicity will be liable and may be subject to sanctions as above indicated.
Click the following links to read more legal articles from Chile:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
9. Preclinical and Clinical Trial Requirements: Chile
1. Are clinical trials required to be conducted locally as a condition (stated or implicit) for marketing approval?
No. Chilean legislation and regulations do not require for clinical trials to be conducted locally in order to file for or obtain a marketing approval, the ISP accepts clinical trials conducted abroad. These studies must demonstrate the safety and efficacy of the product.
2. How are clinical trials funded?
Clinical trials may be privately or publically funded. Good Clinical Practice guidelines, approved by Exempt Resolution N° 460 of the ISP in January of 2015 (hereinafter “GCP guidelines”), defines as sponsor of a clinical trial any individual, company, institution or entity with domicile and legal representative in Chile, responsible of initiating, administrating/controlling and/or financing a biomedical investigation in human beings or clinical study. Additionally, the individual, institution, company or entity that, not financing directly the biomedical investigation, has performed concrete actions to obtain its financing shall also be considered as sponsor.
As set forth under article 99 of the Sanitary Code and under the provisions set forth in Law No. 20.120 –on Scientific investigation on the Human Being, its Genome and Prohibits Human Cloning– and its regulations –set forth in Decree No. 114 of 2011–, to perform a clinical trial in our country, all individuals or entities, whether public, private, national or foreign who are sponsoring the trial with a pharmaceutical product, must request prior approval from the relevant Scientific Ethics Committee, and from the ISP.
3. What are the requirements for preclinical and clinical trial protocols? Who must approve the protocols?
The GCP guidelines follow the guidelines of the ICH Guideline for Structure and Content of Clinical Study Reports. In this regard, GCP guidelines state that clinical trial protocols must generally include the following general content requirements:
- General information: (i) Title of the Protocol, identification number of the protocol and date (any amendment shall also include the number and date of the amendment); (ii) name and address of the sponsor and monitor (if different to the sponsor); (iii) name and title of the persons authorized by the sponsor to sign the protocols and its amendments; (iv) name, title, address and phone number of the medical expert of the sponsor for the study; (v) name and title of the investigators responsible for conducting the study and address and phone number of the site or sites in which the investigation will be conducted. Name, title, address and phone number of the qualified physicians responsible for medical decisions related to the investigation site (if different to the investigator); (vi) Name, address of the clinical laboratories and other medical or technical departments and or sites involved in the study.
- Background information: (i) Summary of the findings in non clinical studies with potential clinical significance and other clinical studies relevant for the investigation; (ii) Summary of known and potential risks and benefits, if available, for human beings; (iii) description and justification of the route of administration, dosage, dosage scheme and treatment schedule; (iv) statement that the study will be conducted in accordance to GCP, the protocol and applicable regulatory requirements; (v) description of the target population of the study.
- Detailed description of the objectives and purpose of the study.
- Study design: (i) description of the primary and secondary evaluation points, if included; (ii) description of the type of study design (e.g. double blind; placebo controlled; parallel) and a schematic diagram of the design procedures and study stages; (iii) description of the measures taken to minimize/avoid bias (e.g. randomization; blinding); (iv) Description of the study treatment, study product dosage and dosage scheme. Also a description of the dosage, packaging and labelling of the study products; (v) Expected time of participation of study subjects and sequence and duration of all periods of the study and study follow-up, if applicable; (vi) description of rules to suspend and discontinue the study, totally or partially and of individual participation of subjects; (vii) procedures for counting the study products including placebo and comparators, if available; (viii) procedures for maintaining codes for randomization and procedures to open codes; (ix) identification of any data to be recorded on the Case Report Forms to be considered as source documents.
- Criteria for Inclusion, exclusion and withdrawal of study subjects (including procedure for withdrawal, collected data, replacement and follow up for withdrawn subjects).
- Subject treatment: (i) name, dosage, dosage scheme; route/means of administration, and treatment period for all treatment to be provided and follow up period for the subjects for each treatment/ study group/study arm; (ii) allowed and non-allowed medication/treatment prior or during the study; (iii) procedures for the monitoring of subject compliance.
- Efficacy evaluation, including specifications of efficacy parameters the methods and timings for their evaluation, recording and analysis.
- Safety evaluation, including specifications of safety parameters the methods and timings for their evaluation, recording and analysis. Additionally, description of procedures to record and generate adverse event reports and intercurrent diseases and follow up of subjects.
- Statistics: (i) statistical methods to be implemented, including planned intermediate analysis; (ii) Number of subjects to be included (per site for multicentric investigations) and the reason for sample size, including study potency and clinical justification; (iii) significance level; (iv) criteria for termination of study; (v) procedure to explain missing; non used or false data; (vi) description of procedures to report and justify any deviation of the original statistical plan ; (vii) study subject selection for analysis.
- Direct access to data/source documents, permitting access, audit and monitoring by the Scientific Ethics Committee and regulatory inspections.
- Quality Control and Quality assurance.
- Ethics considerations for the study.
- Data management and records custody.
- Financing and insurance.
- Publication policy.
Responsibility for the revision and approval of the trial protocols for clinical trial investigation carried out in Chile rely on the relevant Scientific Ethics Committee and additionally by the ISP. If the approved protocol is changed, the Scientific Ethics Committee and the ISP must be notified.
4. What are the requirements for consent by participants in clinical trials?
As per Law No. 20.120 and its regulations, the consent necessary for participants to enter the clinical trial must comply with the following requirements:
- The consent must be granted before the enrollment, shall be a clear, free and informed consent and must be granted in writing.
- The consent may be given by the study subject or by the person authorized by law to provide the consent on behalf of the study subject.
- The consent shall be evidenced in a written form, signed by the study subject, the responsible investigator and by the Director of the Institution (or its delegate) where the trial will be performed, who additionally will attest to the authenticity of the issuance of the consent (ministro de fe).
- The information provided to the subject about the investigation project shall be adequate, sufficient and easy to understand and shall be previously approved by the corresponding Scientific-Ethics Committee as an official document included in the protocol of the scientific investigation.
- Additionally, the subject shall be expressly informed of his/her right to not authorize the investigation or of his/her right to revoke his/her consent at any moment or by any means without any liability, sanction or loss of benefits which will be recorded in the medical record and in the corresponding document of the investigation protocol.
- The consent shall be requested every time the terms and conditions of the investigation are substantially modified (except when such modifications are considered as minor modifications by the Scientific-Ethics Committee).
It is important to note that any mentally disabled individual who cannot express his/her will, shall not be able to participate in scientific investigations. In the case of scientific investigations in which a mentally disabled person who can express his/her will is participating, the approval of the Scientific Ethics Committee is required and, additionally, the approval of the relevant Sanitary Authority, plus the express consent of the study subject and his/her legal representative.
5. May participants in clinical trials be compensated?
Payment or compensation to participants of clinical trials is allowed, as they are there are no provisions under Chilean legislation and regulations prohibiting this kind of compensation.
In this regard, the GCP guidelines indicate that, “neither the investigator nor the staff should oblige, coerce or unduly influence a subject to participate or continue her participation in a study”. The parties involved may determine an amount of compensation as long as such agreement has been fully and clearly informed to the patient. In this regard, the following information must be provided to the participant:
- The pro-rated advanced payment to the subject for participating in the study, if applicable.
- The payment of the anticipated expenses to the subject for participating in the study, if applicable.
Compensation cannot be a donation of pharmaceutical products.
6. How are participants in clinical trials protected and indemnified against any harm that arises as a result of participation in the trial?
Law No. 20.850 on 2015, also known as Ley Ricarte Soto, amended Chilean Sanitary Code regarding clinical trials. This law included a set of rules to specifically regulate circumstances of damages to patients due to their participation in clinical trials.
The holder of the provisional use authorization will be liable for any damage caused during the investigation, even if it could not have been foreseen or prevented according to the state of science at the time the damage occurred, stating that once the damage is proven, it will be presumed that this occurred as a result of the study.
Furthermore, the holder of the provisional authorization must also have civil liability insurance, which is also required for the execution of clinical studies under Law No. 20.120, its regulations and GCP guidelines.
Click the following links to read more legal articles from Chile:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
10. Regulatory, Pricing and Reimbursement Overview: Chile
1. What are the regulatory authorities with jurisdiction over drugs, biologicals, and medical devices in your country?
In Chile the regulatory authority responsible for the enforcement of the regulatory framework for pharmaceutical products, including biologicals, and medical devices is the Public Health Institute (ISP), which is a functionally decentralized and autonomous public service overseen by the Ministry of Health (MoH).
In turn, the Ministry of Health is the main health authority in Chile, which, pursuant to the provisions of the Chilean Sanitary Code, is responsible for the issuance of the respective regulations which govern the import, clearance, export, production, manufacturing, fractioning, storage, handling, transport, distribution, sale, pharmacovigilance, traceability, advertising, promotion or information to professionals, medical use or scientific investigation of pharmaceutical products and for the progressive implementation of the provisions for medical devices.
2. What is the regulatory framework for the authorization, pricing, and reimbursement of drugs, biologicals, and medical devices?
In Chile, the authorization for the commercialization of a pharmaceutical product is governed by the Sanitary Code, the regulations set forth in Supreme Decree No. 3/2010, issued by the MoH, which contains the Regulations for the National Control System of Pharmaceutical Products for Human Use and by ancillary regulations and technical guidelines approved by the MoH and the ISP (e.g. Decree No. 500 of 2012 of the MoH approving Technical Guideline No. 136 establishing the active ingredients included in pharmaceutical products that must demonstrate therapeutic equivalence and its amendments; Decree No. 27 of 2012 of the MoH approving Technical Guideline N° 131 defining the criteria to prove therapeutic equivalence in pharmaceutical products in Chile and its amendments; and Decree No. 945 of 2014 of the MoH approving Technical Guideline No. 170 on sanitary registration for biotechnological products derived from recombinant DNA techniques and its amendments, among several others).
Medical devices are governed by the Sanitary Code and the regulations set forth in Supreme Decree No. 825/1999 which contains the Regulations for Products and Devices of Medical Use. Furthermore, medical devices law and regulations incorporate a progressive implementation through grounded Supreme Decrees issued by the Ministry of Health –prior report issued by the ISP–, indicating the specific medical devices which will need to fulfill the provisions included in the Sanitary Code and Supreme Decree No. 825/99 in order to be manufactured, imported, commercialized and distributed in Chile.
Currently, regulated medical devices to which sanitary restrictions apply include latex surgical gloves for single use, latex medical examination gloves and latex condoms (Decree No. 342/2004 of the MoH), sterile hypodermic needles for single use and sterile hypodermic syringes for single use (Decree No. 1.887/2007 of the MoH) and synthetic masculine condoms and feminine condoms (Decree No. 93/2018 of the MoH).
There is no general regulatory reimbursement process or pricing laws for pharmaceutical products or medical devices.
Nevertheless, the health coverage of pharmaceutical products and medical devices is based on a public and private insurance system and universal coverage programs, being the most relevant the Explicit Health Guarantees (GES plan) and the High Cost Treatment Financial Protection System (Ley Ricarte Soto).
3. What are the steps to obtaining authorization to develop, test, and market a product?
PHARMACEUTICAL PRODUCTS
Any pharmaceutical product, whether imported or manufactured in the country, requires a sanitary registration (marketing authorization) in order to be distributed or used under any title in Chile. A pharmaceutical product may be exceptionally authorized by the ISP to be used temporarily without prior sanitary registration if an epidemic, emergency or catastrophe occurs, or if required for an urgent medical use or for scientific research or clinical trials. We will later provide more information on the provisional use of pharmaceutical products.
In general terms, for the sanitary registration of a pharmaceutical product the applicant will be required to comply with general requirements including the submission of administrative information, technical information, pharmaceutical quality information and data on safety and efficacy of the product. Special requirements will also be applicable for fixed dose combination pro-ducts, pharmaceutical combination products, phytopharmaceutical products; homeopathic products and biologicals.
Safety and efficacy data, including full preclinical and clinical studies for the product will be necessary to be submitted in order to achieve the sanitary registration of a pharmaceutical product under the standard registration procedure (procedimiento ordinario de registro), applicable, in general terms for innovator products. Nonetheless, Chilean regulations, in specific cases, also include the possibility to file for a simplified procedure (procedimiento simplificado de registro), permitting the omission of specific safety and efficacy data, available for generics products, as will be described.
Additionally, during 2020, modifications were made to Supreme Decree No. 3/2010 with the purpose of modifying the conditions for the abbreviated registration procedure and incorporating a new accelerated registration process, as will be described.
3.A. Standard Procedure (“Procedimiento Ordinario”) for the registration of Pharmaceutical Products (new drugs, biologics)
The standard procedure is the general procedure established under Chilean regulations for the sanitary registration of pharmaceutical products and will be applicable in all cases defined under article 53 of Supreme Decree No- 03/2010 which, in general, relates to:
- Pharmaceutical products incorporated for the first time in the field of medicine in Chile;
- For biological products;
- Products containing an active ingredient of a previously registered product granted with regulatory data protection;
- For products including a new therapeutic utility, posology, route of administration or age group of a previously registered product.
- For products which include a modification in the composition and concentration of the active ingredients, or present new salts, esters, isoforms or complexes for an active ingredient of a previously registered product or constitute a fixed dose combination of active ingredients which, separately, may have or not prior sanitary approval.
- Products having a different dosage form of a previously registered product, modifying the release of the active ingredient.
- For the first registration of a combination pharmaceutical product
As indicated above, full preclinical and clinical data on safety and efficacy will be required to be submitted under the standard procedure for sanitary registration of these products.
For biotechnological products, and within the standard procedure of registration, Chilean regulations allow for the abbreviation of clinical data to certify the safety and efficacy provided such product has the same active ingredient, unitary dosage, pharmaceutical form and administration route than another registered reference product. This sets forth the possibility of a biosimilar pathway for specific biotechnological products. The biosimilar pathway, however, is only available for the active ingredients and their respective presentations included in Technical Guideline No. 170 approved by Decree No. 945 of 2014, and its amendments. Technical Guideline No. 170 is based upon the WHO Guidelines for similar biotherapeutic products and structures the biosimilar pathway upon a stepwise comparability on characterization and quality, non-clinical and clinical stages, head to head with the reference product (which is also specifically set within such gui-dance). It also includes provisions in connection to pharmacovigilance and extrapolation of indications.
The administrative steps for the sanitary registration of a product under the standard procedure is described below:
[caption id="attachment_85878" align="aligncenter" width="717"]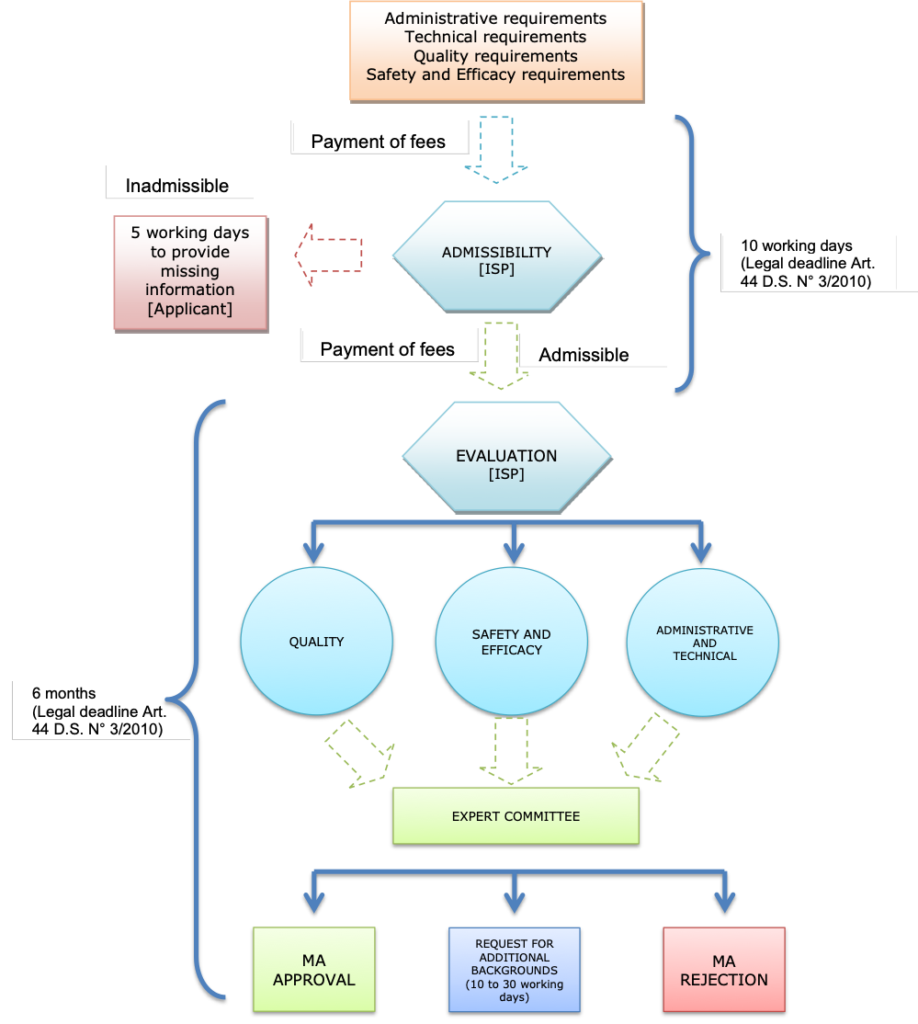 Note: MA= marketing authorization[/caption]
Note: MA= marketing authorization[/caption]
3.B. Simplified Procedure for the registration of Pharmaceutical Products (generics)
The simplified procedure allows for the omission of specific data regarding safety and efficacy of the pharmaceutical product filed for registration and it is permitted, in general, for pharmaceutical products containing the same active ingredient, in the same pharmaceutical dosage form and the same administration route as another product which currently has or had in the past a sanitary registration granted by the ISP and which has not been cancelled for public safety issues.
Specific therapeutic equivalence studies will be required for products containing active ingredients as per Decree No. 500 of 2012 of the MoH approving Technical Guideline No. 136 which establishes the active ingredients included in pharmaceutical products that must demonstrate therapeutic equivalence and its amendments.
The administrative steps for the sanitary registration of a product under the simplified procedure is described below:
[caption id="attachment_85879" align="aligncenter" width="733"]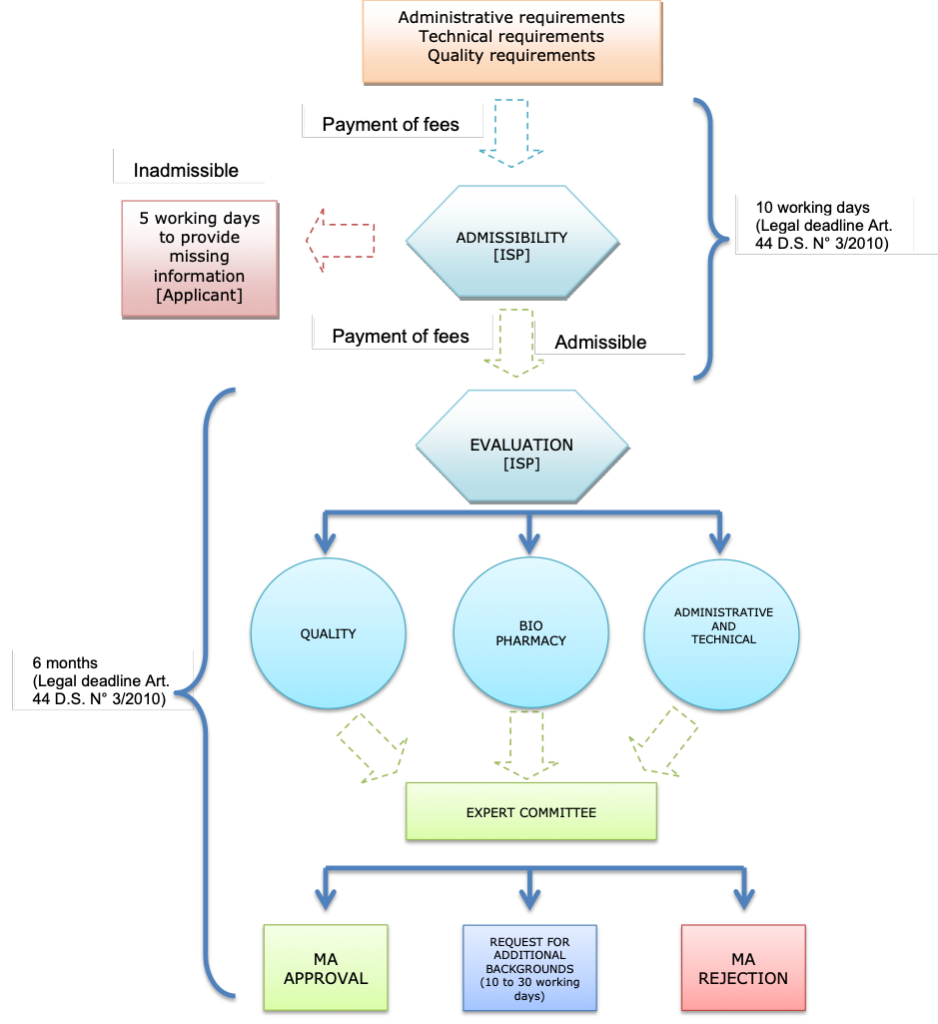 Note: MA= marketing authorization[/caption]
Note: MA= marketing authorization[/caption]
3C. Abbreviated Procedure for the registration of Pharmaceutical Products (abbreviated pathway)
On August 21st, 2020, article 51 of Supreme Decree No. 03/2010 was modified to amend the abbreviated procedure, which now may be declared ex officio by the ISP or at the request of the interested party.
In the event that the abbreviated procedure petition is filed by the interested party, the application can be based on the following circumstances:
- When the pharmaceutical product is needed to be distributed to the population in compliance with plans or health programs approved by the Ministry of Health, in which certain situations of health risk or specific pathologies are addressed and which are destined for certain groups of people, within the framework of national public health interests.
- When the pharmaceutical product has been included in the list of products incorporated in the National Formulary of Medicines, in which case the interested party must use the monographs of the Formulary to speed up the processing of the registration.
According to the regulation, the abbreviated procedure timelines should not exceed four months.
3D. Accelerated Procedure for the registration of Pharmaceutical Products (accelerated pathway)
On August 21st, 2020, article 54 of Supreme Decree No. 03/2010 was also modified to incorporate a new accelerated registration procedure.
This accelerated procedure is based on the products’ approval by High Surveillance Regulatory Agencies, as defined in the provisions, and the approval timeline should not exceed three months since the product is declared admissible, as long as all registration requirements are fulfilled.
To request the accelerated registration procedure, the applicant must indicate in the application the existence of a sanitary registration or authorization for use granted by any of the following regulatory agencies (High Surveillance Regulatory Agencies:
I. Those defined as stringent regulatory authorities in Annex 5 of the “WHO Expert Committee on Specifications for Pharmaceutical Preparations - WHO Technical Report Series, No. 986 - Forty-eighth Report” and its subsequent modifications.
II. Those classified as Level IV in the Evaluation System of National Drug Regulatory Authorities of the Pan American Health Organization.
III. Members of the “Pharmaceutical Inspection Co-operation Scheme” (PIC/S).
In order to obtain the sanitary registration of the pharmaceutical product under the accelerated procedure, the applicant must submit the same supporting information given to the Regulatory Agency that granted the registration (reference agency), request the same therapeutical indication and submit the Certificate of Pharmaceutical Product (CPP) of the reference agency.
The review carried out by the ISP must take into account the revision already performed by the respective reference Agency.
This procedure will not be applicable in the following cases:
- Biological products, unless there is a founded resolution from the Ministry of Health that allows this exception.
- When there are public health reasons regarding a certain pharmaceutical product or category of products, which will be qualified through a founded resolution of the Ministry of Health.
- Products for which the sanitary registration has been denied in one or more High Surveillance Regulatory Agencies.
Provisional Use of Pharmaceutical Products
According to article 99 of the Sanitary Code, the ISP may provisionally authorize the distribution, sale and use of pharmaceutical products without prior registration, for clinical trials or other types of scientific research, as well as for urgent medicinal uses derived from situations of shortage or inaccessibility that may affect people considered individually or collectively.
In this sense, Decree No. 50/2019 amended article 21 of Supreme Decree No. 03/2010 to specify the causes through which the provisions of article 99 of the Sanitary Code would be applied:
- Urgent medicinal use derived from situations of shortage or inaccessibility, affecting people, considered collectively.
- Those pharmaceutical products for urgent medicinal use imported for the exclusive consumption of the importer (for a maximum treatment of 6 months).
- In the case of products to be used in scientific research or clinical trials, with a prior favorable report from the corresponding ethics committee or committees, in accordance with the rules on clinical trials performed on human beings, approved by the Ministry of Health.
In the case of letters a) and b) indicated above, they will be presented to the ISP proving, by any means, the authorization granted by the health authority of the country of origin or manufacture, as appropriate, and the prescription of the pharmaceutical product must be issued by a professional enabled to prescribe, stating the need and duration of the treatment (with a maximum of 6 months of treatment for letter b)). These authorizations may be requested by the interested party as many times as necessary.
MEDICAL DEVICES
Concerning medical devices, as of today, only the following medical devices require a sanitary registration:
- Latex surgical gloves for single use
- Latex gloves for medical examination
- Male natural, rubber and latex condoms
- Female condoms
- Sterile hypodermic needles for single use
- Sterile hypodermic syringes for single use
For regulated medical devices, the ISP will evaluate the product backgrounds based on the risk classification of the medical device.
- Class I: includes devices having a very low level of risk
- Class II: includes devices having a moderate level of risk
- Class III: includes devices having a high risk potential
- Class IV: includes devices considered the most critical in the matter of risk
According to their class, medical devices must comply with quality, safety and efficacy requirements, which will be reviewed by ISP, and certification of the verification of conformity, granted by authorized entities or by the ISP in the absence of them.
All other medical devices, are currently “non-regulated medical devices” which may be imported to Chile and commercialized in the country without further restrictions and require no sanitary registrations or authorization, although they may be subject to voluntary measures.
For regulated medical devices, the applicant must request the “registration” of the medical device before the ISP through filling in form SDM/005 as made available by the ISP, and submitting:
- Certification of the verification of conformity issued by an entity authorized by the ISP.
- Certificate for exporting purposes or free sale certificate issued by the rele-vant sanitary authority, duly legalized.
- Document issued by the manufacturer, where the applicant is recognized as the distributor of its products (mandatory for importers and distributors).
- Certificate of quality management system in the manufacturing process (e.g. ISO 13485; ISO 9001, or others).
To commercialize a regulated medical device it will be necessary to have:
- Authorization of warehouse of storage for medical devices, duly issued by the competent Health Regional Ministerial Secretariat (SEREMI).
- Customs destination certificate, issued by the relevant SEREMI which will permit the transport of the product to the authorized warehouse of the provider.
- Use and Disposition (AUD) resolution, granted by the ISP in order to authorize the use and distribution of controlled medical devices imported to Chile.
- Certification of the verification of conformity of the medical device performed by the authorized entity; and
- Sanitary registration of the medical device in the ISP (as previously explained).
4. What are the approximate fees for each authorization?
Currently, the fees for obtaining a marketing authorization are:| GOVERMENT FEES IN USD FOR MARKETING AUTHORIZATION FOR A: | |
| NEW DRUG (Standard Procedure) | USD 2000 |
| BIOLOGICAL (Standard Procedure) | USD 2000 |
| GENERICS (Simplified Procedure) | USD 1600 |
| GOVERMENT FEES IN USD FOR MEDICAL DEVICES | |
| Registration of the company before ISP | USD 320 |
| Registration of regulated medical device (per product) | USD 100 |
| Certificate of Review of the Antecedents that accompany the non-regulated medical device (per product) | USD 330 |
| Declaration of regulatory status of medical devices (voluntary submission) | USD 25 |
5. For how long are marketing authorizations/registrations valid? How are marketing authorizations/registrations renewed?
Sanitary registrations for pharmaceutical products will last for a period of 5 years, which is renewable for equal and successive periods, if not cancelled.
6. How does the authorization process differ between brand-name products and generic products? Are there differences for local manufacturers versus foreign-owned manufacturers?
Pursuant to article 20 of Supreme Decree No. 03/2010 (S.D. No. 03/2010), every pharmaceutical product, whether imported or manufactured in the country, is required to have a sanitary registration which is provided by the ISP for its distribution or use under any title in Chile. In this regard, there are no differences for local manufacturers or foreign-owned manufacturers.
As stated in question No. 3 above, innovator products shall be subject to the Standard procedure (procedimiento ordinario de registro) and will be required to submit, among other information, the full preclinical and clinical studies to evidence safety and efficacy of the product. For biological products, the generic pathway or simplified procedure for registration is prohibited. Nevertheless, for specific biotechnological products included in Technical Guideline No. 170, a biosimilar pathway is available, permitting the reduction or abbreviation of safety and efficacy data based upon head to head, stepwise comparability studies with the reference biotechnological product in all quality-characterization, non-clinical and clinical stages.
On the other hand, our regulation sets forth the simplified procedure (procedimiento simplificado de registro), which is applicable for generic products. This pathway can be used for pharmaceutical products that have the same active ingredient, in the same amount per pharmaceutical form and the same route of administration as another product that was previously registered. The applicant in this procedure is not required to provide, with the sanitary application, the scientific information related to safety and efficacy required for the standard procedure, unless requested by the ISP by means of grounded resolutions. However, specific therapeutic equivalence studies will be required for products containing active ingredients as per Decree No. 500 of 2012 of the MoH approving Technical Guideline No. 136 establishing the active ingredients included in pharmaceutical products that must demonstrate therapeutic equivalence and its amendments.
7. How are combination products (drug + drug, drug + biologic, drug + device, biologic + device, drug + biologic + device) regulated?
A pharmaceutical combination product is defined under Chilean regulations as a product comprising two or more pharmaceutical products included in a single packaging to be administered sequentially or simultaneously.
For the registration of a pharmaceutical combination product, the applicant must submit all general applicable information as explained in question No. 3 above. Furthermore, Chilean regulations requires for the applicant to prove the safety and efficacy of the proposed combined use of the pharmaceutical products and, additionally, demonstrate:
- That each pharmaceutical product must contribute to the therapeutic effect of the combination product.
- That the dosage of each product, the administration therapeutic scheme and treatment duration must provide safety and efficacy to the combination product, without risk of increasing adverse reactions.
- Compatibility between the ingredients, including excipients, used in each pharmaceutical product from a chemical, pharmacological, pharmacokinetic and biopharmaceutical standpoint, in vitro and in vivo, as applicable.
- That secondary, collateral or toxic effects are of the same or minor intensity as individually shown for each pharmaceutical product.
Pharmaceutical combination products may not include phytopharmaceuticals or homeopathic products, combined among themselves or with other pharmaceutical products.
Combination of devices or devices with pharmaceutical products are not expressly regulated. Nevertheless, the ISP has expressed its criteria in the sense that products which combine a medical device with a pharmaceutical product having only an auxiliary function (e.g. heparin covered catheters), are governed under the regulation for medical devices. On the other hand, products which include a medical device and a pharmaceutical product as a single product (e.g. prefilled syringes) are considered and governed under the rules for pharmaceutical products.
8. How is compliance with regulation monitored and evaluated? Is the regulatory regime comparable with the U.S. Food and Drug Administration or the European Medicines Agency expectations and requirements?
Compliance with regulations is monitored and evaluated by the ISP through periodical inspections, which are previously scheduled by the authority or upon complaints or denouncements received by the authority anonymously or from third parties.
Under the Sanitary Code and applicable regulations, the ISP and other sani-tary authorities are granted with ample faculties to visit, inspect and search any public or private site, to obtain and inspect records and documentation and to impose sanitary measures, applicable upon imminent risk and nece-ssary to safeguard public health. In 2016, the ISP, through Exempt Resolution No. 1290, approved an Inspection Manual for the exertion of these faculties and has also issued internal guidelines for sanitary inspections (available at: http://www.ispch.cl/sites/default/files/manual_interno_fiscalizacion_con_medidas.pdf).
If, within the sanitary inspections performed by the ISP, a sanitary infringement is detected, the authority may open a sanctioning administrative procedure (sumario sanitario), under which it may impose sanctions including fines or other penalties as indicated in question No. 9 below.
9. What is the potential range of penalties for noncompliance?
Penalties and sanctions for non-compliance of sanitary laws and regulations may be imposed by the ISP prior carrying out the respective sanctioning procedure (sumario sanitario). The penalties which it may impose upon finding a sanitary infringement include fines ranging from USD 0.1 to USD 70,000. In case or repeated infringements, previously imposed fines can be duplicated.
Additionally, the ISP can shut down the location or facility where the infringement is committed, prohibit its operation, revoke the permits or authorizations granted, suspend distribution of products, among other sanctions included in section 174 of the Sanitary Code.
10. Is there a national healthcare system? If so, how is it administered and funded?
Yes, there is a national healthcare system mainly formed and structured under Health Services (Servicios de Salud), which are the entities responsible for the execution of actions of promotion, prevention, recovering of health and rehabilitation of patients, besides of enforcing the provisions of the Sanitary Code, when appropriate.
For the developing of the referred actions, they are decentralized state entities, granted with their own legal personality and patrimony. The Assistance Network of each service is formed by the set of public healthcare assistance facilities that are part of the service.
Health coverage is primarily based on a public and private insurance system and two universal coverage programs, which are Explicit Guarantees in Health (GES plan) and a High Cost Treatment Financial Protection System.
Members of the Chilean armed forces (army, navy, air force) have a special social security statute set forth by law No. 18.948 and Law No. 19.465, including healthcare.
The Chilean healthcare system is primarily structured by a mandatory medical coverage which is required by law. This coverage is financed by health insurance contributions paid to the providers of healthcare insurance (FONASA or the ISAPREs as it will be explained in question No. 11) on a monthly basis by certain persons such as employees (in which case the relevant employers are legally bound to withhold the relevant amounts from employees’ monthly wages), independent workers, among others. The law provides for a minimum medical coverage and the additional features depend on the health institution and the health plan chosen by each individual.
11. How does the government (or public) healthcare system function with private sector healthcare?
Under Chilean healthcare regulations, there are basically two types of providers of healthcare insurance: a public insurance, which is the National Health Fund (or “FONASA”) and private insurance, consisting of Health Insurance Institutions (or “ISAPRE”), which are legal entities specifically incorporated for these purposes and authorized by, and subject to the control of, the health authorities.
The affiliation of employees to FONASA is automatic. FONASA has 2 different modalities for health attention access:
- Institutional Attention, where the beneficiaries can access to the health attention granted by the Assistance Network belonging to the National Health Services System (public hospitals).
- Free Choice Modality, reserved to people who contribute with the 7% of their monthly salary and their family, who have the option to choose both the professional and the attention facility, within a list of public and private providers who have signed an agreement with FONASA.
On the other hand, coverage by the ISAPREs must be agreed to through the execution of an affiliation agreement. The objective of the ISAPREs is to grant health benefits to its beneficiaries, whether giving them within their own attention units, or financing them by paying the services provided by clinics, hospitals and other institutions.
However, both health systems are available to the public, and affiliates may choose at any time to join an ISAPRE or to join or return to FONASA. In fact, Chilean Constitution states that all persons “have the right to choose their health system, whether public or private” (art.19 N°9 of the Chilean Constitution). However, there are certain restrictions in connection with the medical coverage which is mandatory to provide by law when changing from FONASA to an ISAPRE or vice-versa (e.g. pre-existing conditions). In general, ISAPREs are only allowed to cease or exclude coverage when permitted by law (e.g. pre-existing conditions, plastic surgery, hospitalization for rest purposes, etc.). In contrast, FONASA cannot exclude coverage.
The amounts that are not covered by the ISAPREs must be directly paid by the relevant beneficiaries (copayment).
In addition to the coverage provided by FONASA and the ISAPREs individuals may choose to hire additional healthcare insurance which is not provided by FONASA or the ISAPREs; but instead by duly registered insurance companies.
12. Are prices of drugs and devices regulated and, if so, how?
Currently, the prices of drugs and devices are not regulated in Chile.13. How are drugs and devices used by patients paid for? What roles do public and private payers play?
Please refer to No. 11 above.14. Who dispenses drugs and devices to patients and how are those dispensers compensated?
Drugs will be dispensed within health entities (hospitals and clinics) or healthcare professionals directly (pharmacist) in the pharmacies. Compensation for drugs and devices will depend on the patient’s health care plan (FONASA / ISAPRE / additional private insurance), coverage under Explicit Guarantees in Health (GES plan) or High Cost Treatment Financial Protection System, if available or through out-of-pocket payment.
15. What are the professional and legal responsibilities of those who dispense drugs and devices? What role do they play in providing patient care, information, and safety?
The professionals who dispense drugs must comply with all regulations applying to reception, storage, transport, distribution and dispensing of drug products. Additionally, they must be in possession of the title of Pharmacist.
According to section 129 A of the Sanitary Code, pharmacies must be under the supervision and technical direction of pharmacist who must supervise the correct dispensing of pharmaceutical products in accordance to the terms of the respective medical prescription and to personally inform a promote rational use of medicines, responding questions which patients may have. They shall also exert a permanent supervision of sanitary and technical aspects of the site, irrespective of the responsibility over administrative operation of the site which shall oversee the pharmacy staff.
Supreme Decree No. 466 of 1984 of the MoH, which sets forth the regulations for pharmacies, wholesalers, pharmaceutical warehouses, cabinets and authorized deposits, further complements such legal responsibilities for professionals authorized for the dispensing of pharmaceutical products. In this regard, the technical director of the facilities, must verify and control the adequate dispatch of medical prescriptions assuring compliance with the conditions of sale approved for every product, assuring the correct storage conditions for pharmaceutical products are observed and training and supervising the pharmacy staff in their functions and in compliance with sanitary provisions, among others.
Click the following links to read more legal articles from Chile:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs